Multiplexed Barcoding Image Analysis for Immunoprofiling and Spatial Mapping Characterization in the Single-Cell Analysis of Paraffin Tissue Samples
Summary
Multiplexed barcoding image analysis has recently improved the characterization of the tumor microenvironment, permitting comprehensive studies of cell composition, functional state, and cell-cell interactions. Herein, we describe a staining and imaging protocol using the barcoding of oligonucleotide-conjugated antibodies and cycle imaging, which allows for the use of a high-dimensional image analysis technique.
Abstract
Multiplexed imaging technology using antibody barcoding with oligonucleotides, which sequentially detects multiple epitopes in the same tissue section, is an effective methodology for tumor evaluation that improves the understanding of the tumor microenvironment. The visualization of protein expression in formalin-fixed, paraffin-embedded tissues is achieved when a specific fluorophore is annealed to an antibody-bound barcode via complementary oligonucleotides and then sample imaging is performed; indeed, this method allows for the use of customizable panels of more than 40 antibodies in a single tissue staining reaction. This method is compatible with fresh frozen tissue, formalin-fixed, paraffin-embedded tissue, cultured cells, and peripheral blood mononuclear cells, meaning that researchers can use this technology to view a variety of sample types at single-cell resolution. This method starts with a manual staining and fixing protocol, and all the antibody barcodes are applied using an antibody cocktail. The staining fluidics instrument is fully automated and performs iterative cycles of labeling, imaging, and removing spectrally distinct fluorophores until all the biomarkers have been imaged using a standard fluorescence microscope. The images are then collected and compiled across all the imaging cycles to achieve single-cell resolution for all the markers. The single-step staining and gentle fluorophore removal not only allow for highly multiplexed biomarker analysis but also preserve the sample for additional downstream analysis if desired (e.g., hematoxylin and eosin staining). Furthermore, the image analysis software enables image processing-drift compensation, background subtraction, cell segmentation, and clustering-as well as the visualization and analysis of the images and cell phenotypes for the generation of spatial network maps. In summary, this technology employs a computerized microfluidics system and fluorescence microscope to iteratively hybridize, image, and strip fluorescently labeled DNA probes that are complementary to tissue-bound, oligonucleotide-conjugated antibodies.
Introduction
The tumor microenvironment (TME) is extremely heterogeneous, consisting of tumor cells, tumor stromal cells, immune cells, noncellular components of the extracellular matrix, and numerous abundant molecules produced and released by tumor, stromal, and immune cells1,2. Accumulating evidence demonstrates that the TME has a pivotal role in reprogramming tumor differentiation, growth, invasion, metastasis, and response to therapies3.
Understanding how different cell types in the TME interact and communicate with each other through signaling networks is essential for improving cancer diagnosis, optimizing immunotherapy, and developing new treatments4. Traditional tissue microscopy techniques, including immunohistochemistry (IHC) and immunofluorescence (IF), have been used for decades to study the cell types, abundance, and communications in tumor samples. Unfortunately, these techniques typically can evaluate only one or two protein markers in a tissue section and cannot reveal the complex spatial and structural relationships among these cells5,8,7.
Over the past two decades, several multiplexed imaging technologies have been established8. These technologies provide much improved views of the composition, function, and location of immune cells within the TME, leading to rapid advancements in the ability to identify and spatially profile complex TMEs at the single-cell level9,10. The spatial and structural relationships of various tumor and immune cells in the TME are now at the forefront of biological and clinical studies using these multiplexed imaging technologies11,12.
The recently developed multiplexed imaging technology using oligonucleotide-conjugated antibody barcoding is an influential single-cell biological research platform based on the detection of oligonucleotide-conjugated antibodies in formalin-fixed, paraffin-embedded (FFPE) samples13,14. Currently, this multiplexed imaging technology allows for the simultaneous imaging of more than 100 markers in a single tissue section15, which has increased the number of cell types that are distinguishable in situ. This enables a level of spatial analysis of tumor and immune cells that is not possible using traditional immunophenotyping approaches16.
Herein, we describe an optimized protocol for conjugating purified antibodies to oligonucleotides and validating this conjugation using the multiplexed imaging platform and a multicycle imaging procedure with FFPE tissue. In addition, we describe the basic image processing and data analysis procedures used with this technology.
Protocol
This retrospective study was approved by the Institutional Review Board of the University of Texas MD Anderson Cancer Center. The FFPE tissue samples were collected from patients at MD Anderson as part of routine standard care. No diagnostic or therapeutic interventions were performed. Informed consent was obtained from the patients for using the samples collected for research and publication.
1. Antibody sources used for the antibody panel design
- Create an antibody panel for multiplexed imaging after carefully considering the quality of the tissues and proteins of interest. Three sources of antibodies are considered for the antibody panel design: 1) fully commercially validated antibodies, 2) multiplexed imaging technology-screened antibodies, and 3) end user-shared antibodies.
NOTE: Multiplexed imaging technology-screened antibodies have been shown to work and are available from vendors. These screened antibodies can be applied to multiplexed image staining after oligonucleotide conjugation by the user. - If a protein of interest cannot be found in the sources mentioned above, employ the antibody clones that are known to work with IHC. In addition, IgG isotypes rather than IgM clones are recommended for this multiplexed imaging technology owing to the higher failure rate with IgM than with IgG clones.
2. Before antibody conjugation
- When identifying antibody clones for conjugation using multiplexed imaging barcodes, consider purchasing carrier-free antibodies in phosphate-buffered saline (PBS) or a similar buffer. Carrier proteins, including BSA, gluten, glycerol, and other protein additives, are known to reduce the conjugation capacity.
- Select the most suitable antibody clone, and always optimize the staining conditions (i.e., antigen retrieval and titration) before conjugation. Do this by applying an unconjugated antibody clone to positive and negative tissue for this antibody using standard IF or IHC.
NOTE: If a purified antibody is not commercially available, an antibody purification process must be performed before conjugation. The purification procedure used with antibody purification kits is not discussed herein.
3. Antibody conjugation
- Obtain the antibody conjugation reagents. Commercially available conjugation kits contain a filter-blocking solution, reduction solution 2, conjugation solution, purification solution, antibody storage solution (all stored at 4 °C), and reduction solution 1 (stored at −20 °C).
- Conjugation
NOTE: A purified antibody is treated with a reducing agent, allowing the reduced moieties of the antibody to react with the multiplexed imaging barcode used with this technology and, thus, form a covalent bond. This process takes about 4.5 h and results in roughly 120 μL of conjugated antibody, which is viable for 1 year. All the spin-down is conducted at room temperature (RT), and the flow-through is discarded except in the very last step (3.2.9), in which a collection tube contains the conjugated antibody.- Aspirate and apply 500 μL of the filter-blocking solution using a pipette to the 50 kDa molecular weight cutoff filter columns, and spin down at 12,000 x g for 2 min to block nonspecific antibody binding.
NOTE: If residual solution at the top of the column is noticeable, invert the filter in the collection tube, and spin down at 3,000 x g for 2 min. - Pipette 50 μg of the antibody in a 100 μL volume of solution to the filter column, and spin down at 12,000 x g for 8 min.
NOTE: Use a spectrophotometer to measure the concentration of the purified antibody and to calculate the volume of solution corresponding to 50 μg of the antibody. If the volume of the antibody solution is less than 100 μL, adjust the volume to 100 μL by adding 1x PBS. Retain 1 μg of unconjugated antibody for conjugation confirmation (step 4.4). - Pipette 260 μL of reduction master mix (20 μL of reduction solution 1 mixed with 825 μL of reduction solution 2, which is sufficient for three antibody conjugation reactions) to each filter column. Gently vortex the master mix for 2-3 s, or pipette up and down to mix the solution with the antibody. Incubate at RT for 30 min.
- After incubation, spin down the filter columns at 12,000 x g for 8 min. Add 450 μL of conjugation solution to the filter columns, and spin down at 12,000 x g for 8 min.
- Resuspend the desired barcode in 10 μL of nuclease-free water and 210 μL of conjugation solution.
NOTE: Prepare an antibody tag immediately prior to use for the staining. Do not reuse antibody barcode aliquots. - After the completion of the spin-down in step 3.2.4, add the resuspended antibody tag (approximately 220 μL) into each corresponding filter column. Pipette the mixture up and down gently to mix the reagents. Close the filter column lids, and incubate the conjugation reaction at RT for 2 h. After 2 h, spin down the filter columns at 12,000 x g for 8 min.
NOTE: Setting aside 5 μL of the conjugated solution in a polymerase chain reaction tube and storing it at 4 °C are recommended for the confirmation protocol (see below). - Pipette 450 μL of the purification solution into each filter column, and spin down at 12,000 x g for 8 min. Repeat three times.
- Pipette 100 μL of the storage solution into the filter columns. Gently pipette the mixture up and down more than 10 times, and carefully wash the sides of the filters in the column.
NOTE: Dissolve 50 µg of antibody in 100 μL of the storage solution. If the conjugation reaction is started with more than 50 μg of antibody, add more storage solution at this ratio. - Invert the filter columns in a fresh collection tube. Spin down at 3,000 x g for 2 min at RT. Keep the collected solution. Pipette the conjugated antibody solution into sterile screw-top tubes, and store for up to 1 year at 4 °C.
NOTE: A conjugated antibody should be tested with the multiplexed imaging technology after 2 days. Testing prior to this may result in high background nuclear staining.
- Aspirate and apply 500 μL of the filter-blocking solution using a pipette to the 50 kDa molecular weight cutoff filter columns, and spin down at 12,000 x g for 2 min to block nonspecific antibody binding.
4. Conjugation confirmation
NOTE: Before performing staining experiments with a user-conjugated antibody using the multiplexed imaging technology, the conjugation should be confirmed by using gel electrophoresis with 5 μL of the conjugated antibody (see step 3.2.6) along with 1 μg of an unconjugated antibody (usually in 2 μL of the mixture) as a control. A successful antibody conjugation will be demonstrated by an increase in the molecular weight of the antibody. However, this confirmation protocol only assesses the success of the chemical reaction for conjugation and does not address the antibody validation used for multiplexed imaging.
- Pipette 8 μL and 11 μL of nuclease-free water into the reserved conjugated antibody and control unconjugated antibody, respectively, to obtain a final volume of 13 μL.
- Pipette 5 μL of LDS (or other sodium dodecyl sulfate-polyacrylamide gel electrophoresis system) sample buffer and 2 μL of sample-reducing agent into each sample of the reserved conjugated antibody, and denature the samples in a 95 °C dry bath for 10 min.
- While the samples are denaturing, dilute 40 mL of MOPS SDS running buffer in 760 mL of ultrapure water, place a gel in the tank of an electrophoresis system, and pour the diluted running buffer over the gel.
- Once the 10 min denaturization period is complete, load one well of the gel with a pre-stained protein standard, one with the unconjugated antibody (from step 3.2.2), and the remaining wells with the conjugated antibody samples. Next, run the gel at 150 V until the protein standard appears at the end of the gel.
NOTE: The gel easily adheres to microwave-safe containers and, thus, may tear, so the gel must be handled with caution in the following steps. - After the run is complete, transfer the gel to a microwave-safe container prefilled with ultrapure water, and heat it in a microwave until the first bubble in the water is visualized.
NOTE: The time for bubbles to form varies greatly depending on the microwave used. - Drain the water from the container, pour about 250 mL of Coomassie G-250 stain over the gel, and heat the gel in a microwave until the first bubble is visualized. Afterward, remove the container with the gel and the Coomassie G-250 stain from the microwave, and place it on a shaker for 10 min.
- After shaking, carefully drain the stain, replace it with about 200 mL of ultrapure water, and then place the container on the shaker to wash the gel.
- Drain the ultrapure water, and replace it with new ultrapure water five times or until the remnants of the stain are not apparent in the water bath. Leave the gel to wash overnight on the shaker until bands are visible, if needed, before photographing the gel (Figure 1).
NOTE: The antibodies used in multiplexed imaging should have staining patterns comparable with those of dye-conjugated antibodies. Tissue sections with known antigens positive for conjugated antibodies can be stained with oligonucleotide-conjugated and dye-conjugated antibodies. In this work, in each case, the tissue morphologies for both antibody types were equivalent and harmonized with each other, as well as the anticipated cell distribution, based on the biology of the proteins of interest and the test tissue samples. This result demonstrates the effectiveness of using oligonucleotide-conjugated antibody moieties for tissue staining-based approaches using FFPE tissue.
5. Oligonucleotide-conjugated antibody staining
- Prepare coverslips for tissue placement.
- Soak the coverslips in a 0.1% poly-L-lysine solution for 24 h at RT to improve the tissue adherence.
- After soaking, drain the poly-L-lysine solution, and wash the coverslips with ultrapure water for 30 s. Repeat the washing four to six times. Remove the coverslips from the ultrapure water used for washing, and place them on a lint-free towel to dry overnight.
NOTE: A histologist must cut the selected tissue into sections that are 5 μm thick, place them on the center of a poly-L-lysine-charged coverslip, and allow them to dry overnight. After drying, the coverslips with the tissue sections must be stored at 4 °C for no longer than 6 months. The staining panel in this protocol contained 26 markers. The tissue used in this protocol was normal human tonsil tissue. The coverslip soak time should not exceed 1 week. Poly-L-lysine-coated coverslips may be stored at RT and must be used within 2 months of preparation.
- The day before staining, place the coverslip holder in a 60 °C oven overnight.
- The following day, place the coverslip sample in a preheated coverslip holder in a 60 °C oven. After baking for 30 min, check to ensure that the paraffin has melted away from the tissue.
- Quickly place the coverslip holder/sample in the following solution series: two rounds of dewaxing agent for 6 min each; two rounds of 100% ethanol for 5 min each; one round of 90% ethanol for 5 min; one round of 70% ethanol for 5 min; one round of 50% ethanol for 5 min; one round of 30% ethanol for 5 min; and two rounds of diethyl pyrocarbonate (DEPC)-treated ultrapure water for 5 min each.
NOTE: All the dilutions of ethanol are prepared with DEPC-treated ultrapure water. If xylene is used for dewaxing, it should be used in a hood. - While subjecting the coverslip holder/sample to the solution series, do the following:
- Prepare a humidity chamber by placing an empty pipette tip box with a water-soaked paper towel at the bottom.
- Fill a pressure cooker with enough water to halfway cover a 50 mL beaker.
- Place 5 mL of methanol per sample in the beaker at 4 °C.
- Dilute AR9 buffer with DEPC-treated ultrapure water to 1x; about 50 mL of the diluted buffer is needed per coverslip holder.
- After the solution series is complete, fill a 50 mL glass beaker with about 40 mL of 1x AR9 buffer, submerge the coverslip holder/sample in the beaker, and completely cover the beaker/coverslip holder with aluminum foil.
- Place the aluminum foil-covered beaker/coverslip holder in the water-filled pressure cooker, and cook at high pressure (about 15 psi) for 20 min.
- After cooking, remove the beaker/coverslip holder, carefully unwrap the aluminum foil, and allow the beaker/coverslip holder to cool at RT for about 10 min.
- Remove the coverslip holder/sample from the 1x AR9 buffer, and submerge it in two rounds of DEPC-treated ultrapure water, performing an incubation of the samples in both rounds for 2 min each.
- During the incubation, retrieve the hydration buffer, the blocking N, blocking G, blocking J, and blocking S solutions, and the antibody diluent/block from the multiplexed imaging staining kit. For two coverslip samples, label 6-well plates for the solutions using the configurations shown in Figure 2.
- Place the coverslip sample in two rounds of 5 mL of the hydration buffer for 2 min each.
- After both rounds of placement in the hydration buffer, place the coverslip sample in 5 mL of the multiplexed imaging antibody diluent/block, and incubate for 20-30 min at RT (do not exceed 30 min).
- During the incubation, prepare an antibody cocktail by making a 200 μL master mix of multiplexed imaging antibody diluent/block, N blocker, G blocker, J blocker, and S blocker solutions.
NOTE: Calculate the total antibody amount based on the number of markers and each marker's validated titration, and subtract the total antibody amount from the master mix. For example, for six markers, each with a 1:200 titration, the equation would be 200 μL of master mix − 6 μL of antibody cocktail = 194 μL of master mix.
- After incubation in the multiplexed imaging antibody diluent/block, place the coverslip sample in the humidity chamber prepared in step 5.5.1, pipette 190 μL of the antibody cocktail onto the coverslip sample, and incubate the coverslip sample at RT for 3 h.
- After incubation, wash the coverslip sample in two rounds with 5 mL of the multiplexed imaging antibody diluent/block for 2 min each.
- To fix the bound antibodies to the tissue on the coverslip, perform steps 5.7.3-5.7.5 in the humidity chamber.
- Incubate the coverslips for 10 min with 16% formaldehyde diluted to 1.6% with storage solution, and then wash the coverslips three times with 1x PBS.
- Incubate the coverslips for 5 min with 4 °C methanol, and then wash the coverslips three times with 1x PBS.
- Incubate the coverslips for 20 min with 5 mL of the fixative reagent diluted with 1x PBS, and then wash the coverslips three times with 1x PBS.
- Store the stained coverslip samples in the storage buffer at 4 °C for up to 2 weeks
6. Multiplexed imaging reporter plate
NOTE: A 96-well plate, referred to as a reporter plate, containing barcoded fluorophores in individual wells is prepared according to custom-designed multiplexed imaging experiments and correlates with each stained coverslip sample. The following steps are for the preparation of the reporter plate.
- Prepare a reporter master mix by combining 4,880 μL of nuclease-free water, 600 μL of 10x multiplexed imaging buffer, 500 μL of an assay reagent, and 20 μL of nuclear stain solution. This will be enough for 20 cycles of all the wells.
- In every cycle of the custom-designed multiplexed imaging experiment, fill a well with 245 μL of a solution containing the reporter master mix and the specific barcoded fluorophores for that cycle.
NOTE: See Figure 3 for the reporter plate configuration used in this protocol for a carcinoma panel. - To protect the barcoded fluorophores, adhere a foil plate cover over the reporter plate, and place the plate within the multiplexed imaging instrument.
- Store the reporter plate in a dark box at 4 °C for up to 2 weeks.
7. Calibrating and running the multiplexed imaging machine
NOTE: The high-resolution imaging fluorescence microscope captures four different fluorescence channels in each multiplexed imaging cycle at 20x, 100% excitation light, and with low photobleaching.
- Calibrate the focus of the imaging using the DAPI channel by placing a sample coverslip on the microscope stage, manually pipetting 700 μL of a 1:1,500 titration of nuclear stain solution onto the tissue.
NOTE: The coverslip is retained on the microscope stage during the sample washing and imaging. - To prepare the multiplexed imaging instrument, dilute 10x multiplexed imaging buffer to 1x using DEPC-treated ultrapure water, and fill reagent bottles with appropriate solutions/solvents, including the diluted 1x multiplexed imaging buffer, DEPC-treated ultrapure water, and dimethyl sulfoxide (DMSO).
- Once the reagent bottles are filled appropriately, enter the experimental design into the multiplexed imaging instrument manager software; designate the correct cycle, well numbers, z-stack places, marker name, class, and exposure time for each cycle (Figure 4); set all the microscope parameters; and select the regions of interest on the sample coverslip to be imaged.
- Click on the Experiment button in the control software (Figure 4A). In the Experiment Setup and Management window, click on the New Template button (Figure 4B).
- Type the project name into the space next to the Project button (Figure 4C). Type or select the total number of cycles (Figure 4D).
- Click on the Channel Assignment button, type the information for each cycle into the columns (Figure 4E), and click on the Save Template button. Start the experiment by clicking on the Start Experiment button.
NOTE: During a multiplexed imaging experiment, the instrument retrieves the barcoded fluorophores from one well of the reporter plate (maximum of four fluorophores per well, including DAPI), dispenses them directly onto the sample coverslip, and images the regions of interest for each fluorescence channel. Following all the imaging for that cycle, the instrument washes the barcoded fluorophores off and dispenses the next cycle of reporters (from the next well on the reporter plate). Imaging continues until all the cycles using 26 markers have been completed.
8. Image collection
NOTE: Multiplexed images can be collected using any adapted inverted fluorescence microscope configured with four fluorescence channels (DAPI, Cy3, Cy5, and Cy7) and equipped with a Plan Fluor 20x lens. Imaging and washing of the coverslip samples are iteratively performed automatically using a specially developed fluidics setup. The images are acquired using Processor software (v1.8.0.7) in QPTIFF format.
- In the processor window, click on the Input button, and select the experiment name.
- In the Processing Options section, select and check Background Subtraction, Deconvolution, Extended Depth of Field, and Shading Correction. Click on the Start button.
9. Image analysis
NOTE: The acquired images can be uploaded to a patented automated image analysis software or open-source software program (Figure 5) for downstream analysis.
- Click the QuPath icon on the computer, and open the software. Drag the QPTIFF file to the Viewer window.
- Click on the Brightness & Contrast button, which will open the Brightness & Contrast window. Check or uncheck the marker in the Selected column to show or close the marker signal.
- Click on the Zoom to fit button to zoom in or out of the area of interest.
NOTE: Multiplexed image data visualization provides the user with a deep look into the tissue microenvironment. Individual markers in FFPE samples can be visualized in fluorescence format (Figure 6 and Figure 7) or in pathology view. Several computational platforms can be used to process the composite images and analyze the multiplexed tissue image data. Using this software, spatial phenotyping, as well as rare cell discovery and cell neighborhood computation, can be accomplished with whole-slide images of ultra-high multiplexed tissue generated via the multiplexed imaging technology. See Figure 6 for the 26 antibodies in the carcinoma panel and tonsil sample (Supplementary Figure 1).
Representative Results
We employed FFPE tonsil samples to develop a 26 marker immune oncology panel to illustrate the immune status of FFPE tissue using a barcoding image analysis system. Overall, 19 antibodies are currently used in other multiplexed imaging studies in our lab. All of the markers have been tested using FFPE tissue with chromogenic IHC. All the antibodies were conjugated to unique DNA oligonucleotides. When setting up the coverslips using the web-based instrument manager (Figure 4) for this barcoding image analysis technology, it should be noted that the first and last cycles are always "blank" (Figure 2 and Figure 4), which provides the background fluorescence signals to be subtracted from the specific signals from the antibodies. After images in QPTIFF format have been collected with the fluorescence microscope, they can be visualized using several patented automated image analysis software or open-source software programs. Composite images can show all the markers or selected markers for a better view of the signals (Figure 5). Moreover, each antibody can be visually evaluated for nuclear, cytoplasmic, or membranous localization. Immune, tumor, and stromal cells can be easily identified. Subsequently, image analysis can provide information on the signal intensity, dynamic range, and spatial distribution of all the markers (Figure 6). This technique allowed us to analyze all 26 markers at the subcellular level in a single tissue section (Figure 7). By analyzing the markers' co-localization, we could identify the cellular phenotypes, localize the spatial cell position, calculate the distance between cells, and find the distribution of the cells. The crucial impact of this technology is the presentation of a robust 26 marker panel focused on the immune status of the tissue microenvironment.
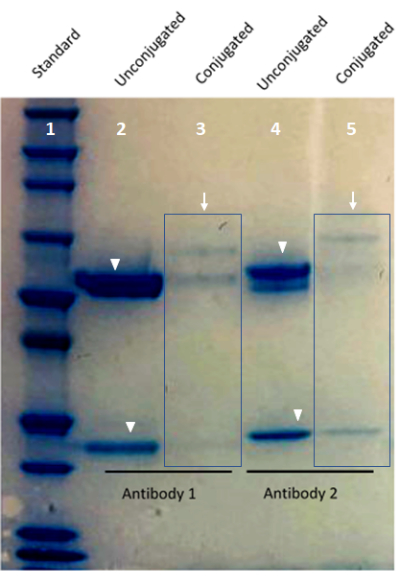
Figure 1: Image of custom-conjugated antibody validation using a Bis-Tris protein gel. Lane 1 of the gel shows the protein standard. Lane 2 and Lane 4 show barcode-conjugated antibodies (arrows). Lane 3 and Lane 5 show the heavy and light chain bands from an unconjugated antibody (arrowheads). Please click here to view a larger version of this figure.
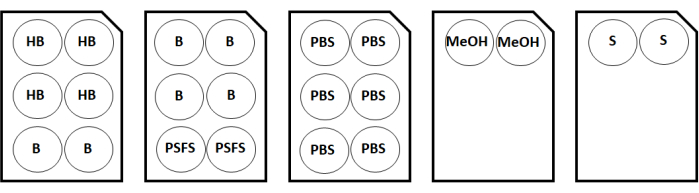
Figure 2: Staining plate configuration map for two coverslip samples. Abbreviations: HB = hydration buffer; B = antibody diluent/block; PSFS = post-staining fixative solution; PBS = phosphate-buffered saline; MeOH = methanol; S = storage solution. Please click here to view a larger version of this figure.

Figure 3: Reporter plate configuration. Each conjugated antibody has a barcode that is complementary to a specific reporter. To set up the reporter plate, every conjugated antibody and its corresponding reporter should be listed. Next, each antibody is assigned a cycle number. The performance of two blank cycles (C1 and C18) is used to evaluate the level of autofluorescence in the three fluorescence channels and for post-imaging background subtraction using the image acquisition control software. A software wizard will check the instrument at this stage to ensure all the settings are correct (Figure 4). Please click here to view a larger version of this figure.
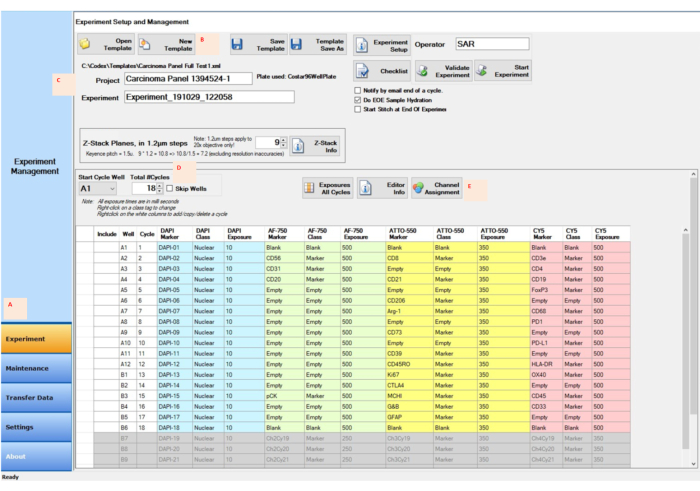
Figure 4: Image acquisition using the control software for the experiment setup. (A) Select the Experiment tab in the left-bottom corner of the control software to prepare and start the setup. (B,C) Select New Template to input the experimental settings with a new project and experiment name. (D) Change the start cycle well and the number of cycles to reflect the reporter location in the 96-well reporter plate. (E) Assign the proper fluorescence channels to the four channels designated for the experimental run. Please click here to view a larger version of this figure.
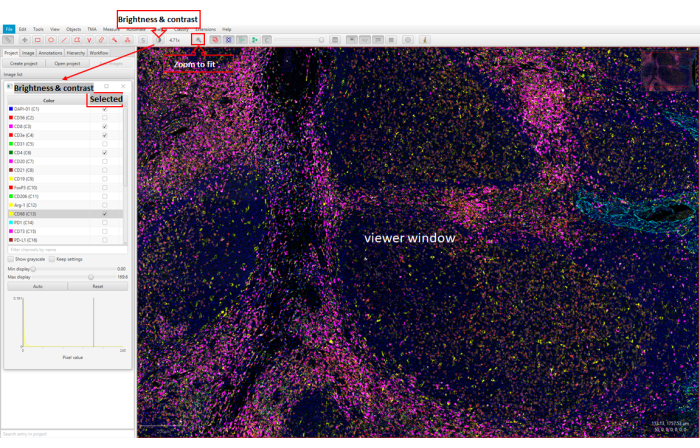
Figure 5: Image visualization using web-based software (QuPath). The viewer window shows 26 markers in the stained tonsil tissue FFPE section. The Brightness & Contrast window shows the markers with the checkmarks. Finally, the viewer window shows the FFPE sample with the selected markers. Please click here to view a larger version of this figure.
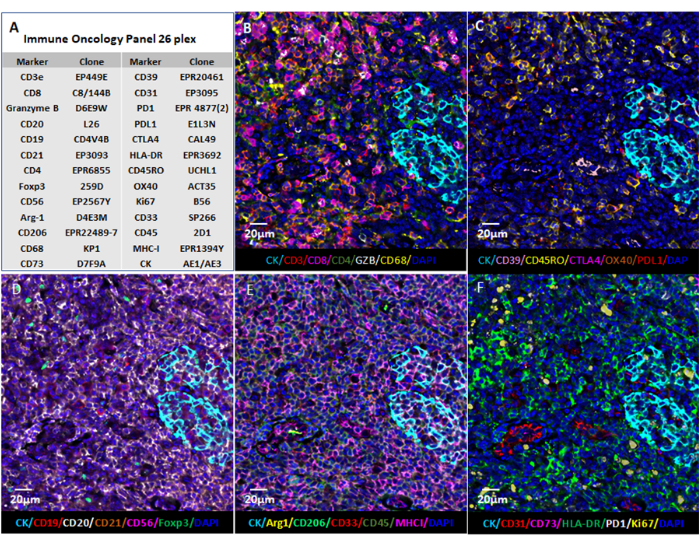
Figure 6: Image visualization using web-based software. (A) Tonsil tissue sections were stained for the 26 markers, and the images of the sections in QPTIFF format were visualized using commercial digital slide viewer software or open-source software (QuPath) for annotation and review. (B–F) Six markers were displayed in the same annotation for a better view of the signals. Please click here to view a larger version of this figure.
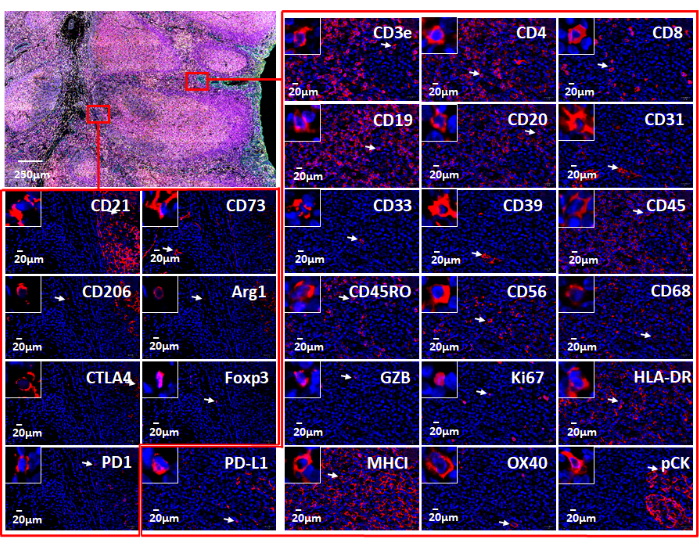
Figure 7: Views of the 26 individual markers used with web-based software. The marker expression in the tonsil tissue is shown via immunofluorescence staining with an immune oncology panel (top left). Individual markers in two small areas (red rectangles) are shown. The zoomed-in insert shows cells positive for these markers (white arrows). Please click here to view a larger version of this figure.
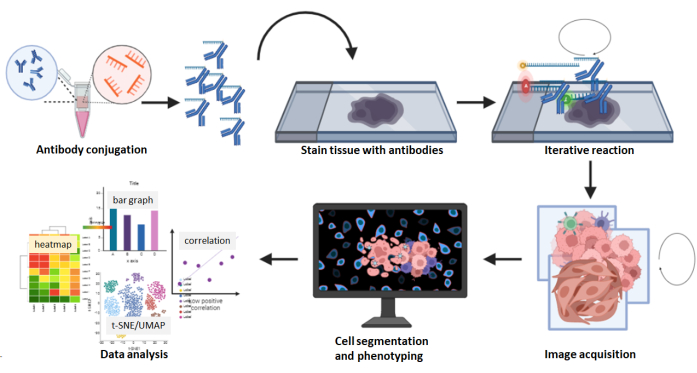
Figure 8: Summary of the multiplexed image acquisition workflow. FFPE tissue sections were stained using the 26 antibody panel followed by a multicycle reaction. Raw images of the stained sections were computationally processed, and a cell density and spatial analysis was performed using the composite images. Please click here to view a larger version of this figure.
Supplementary Figure 1: IHC validation in tonsil tissue. FFPE tissue sections were stained using an individual antibody. The marker expression in the tonsil tissue is shown at a low magnification, and the zoomed-in insert shows cells positive for the marker (red rectangles). Please click here to download this File.
Discussion
The TME plays an essential role in cancer development, progression, and treatment responses. Additionally, the density of specific tumor-infiltrating lymphocyte subsets in the TME can serve as a prognostic biomarker for certain types of cancer. Remarkably, in addition to the TME's cellular composition, the spatial characteristics of a tumor can provide an outline for understanding the tumor's biology and identifying potential prognostic biomarkers12,17.
As numerous immune cell populations are involved in procancer or anticancer responses, a better understanding of these cells and their spatial relationships with each other and with cancer cells will help guide the identification of new immunotherapeutic strategies. Previous studies have stratified the location and spatial distribution of TME cells based on the tissue structure in the intratumoral and peritumoral areas and the invasive margins of the tumor cells18,19. Over the past 15 years, technological advancements have made the phenotypic analysis of individual cells based on their spatial dispersal a novel, influential tool for studying the TME and categorizing potential biomarkers for tumor immunotherapy. Multiplex IF histochemistry can concurrently estimate multiple biological markers20.
Similar to the oligonucleotide-conjugated antibody strategy, four types of protein-based multiplex platforms are used to study the TME: chromogen-, fluorescence-, DNA barcode-, and metal isotope-labeled antibody detection systems. The cost-effective chromogenic IHC platforms enable whole-slide visualization and pathological assessment by using conventional bright-field microscopy. In multiplexed IF and IHC, antibodies conjugated with fluorophores are used. The multiplex IF/IHC platform detects antibodies with high specificity and can quantify targeted antibodies even at the subcellular level6,21. In addition, owing to the nature of chromogens and fluorophores, the use of one antibody panel can capture the expression of up to 10 biomarkers on a single slide. On metal isotope-based platforms, metal-tagged antibodies are used to perform multiplexed imaging with single-cell and spatial resolution, and high sensitivity for individual tissue sections22. Theoretically, these metal-conjugated antibody approaches enable the simultaneous detection of more than 100 biomarkers on a single tissue section. One challenge of the isotope-labeling technique is isobaric interference, which prevents 100% purity of enrichment from being reached23. Moreover, the interference increases as the number of markers increases. DNA-conjugated antibody detection platforms recognize antibodies labeled with unique DNA barcodes. More than 40 biomarkers can be simultaneously captured with high specificity on these platforms6.
Multiplexed imaging is a commercially available DNA barcode-labeled antibody detection platform for applying DNA-conjugated antibodies to a single tissue slide in one step (Figure 8). For the tissue preparation stage, unlike the multiplexed ion beam imaging platform, which requires the use of gold-coated slides obtained from manufacturers, the multiplexed imaging platform requires only regular coverslips or slides coated with 0.1% poly-L-lysine to help the tissue adhere to it and keep tissue intact during the staining and imaging process. The use of tissue sections on coverslips within 4 weeks after sectioning is recommended, as the prolonged storage of unstained slides results in antigenicity reduction. A stained coverslip sample can be maintained in storage buffer at 4 °C for up to 2 weeks without losing its staining signal. No special equipment is required for the storage of the coverslip samples. The multiplexed imaging system has been upgraded to use regular slides instead of coverslips, which enables the staining of larger tissues and easy handling. When using a reduction solution for antibody conjugation (step 3.2.3), the reaction should be limited to no more than 30 min to prevent damage to antibodies. The blocking buffers in step 5.6.6 should be freshly prepared, and the blocking buffers must not be reused.
Compared with chromogen-, fluorescence-, and metal isotope-labeled multiplex antibody detection platforms, the multiplexed imaging technology has certain advantages. For example, more than 60 predesigned antibody panels for multiplexed imaging are commercially available, which helps save time and costs in antibody conjugation and validation, and the number of predesigned antibody panels is growing. These antibodies, which include the carcinoma marker pan-cytokeratin, the melanoma marker SOX10, the vascular marker CD31, the stromal marker SMA, and numerous immune cell markers, are validated and experiment-ready. For antibodies that are not predesigned, the commercially available conjugation kit designed for use with multiplexed imaging is straightforward and user-friendly. Customer-conjugated antibodies are good for 1 year when stored at 4 °C. Additionally, machine warm-up is not required for capturing the images. In this multiplexed imaging technology, the iterative washing, hybridization, and stripping steps in the image acquisition rarely result in decreased marker intensity or degraded tissue morphology5,24,25. Furthermore, composite images are captured in QPTIFF format with a simple three-color fluorescence microscope and can be uploaded and analyzed using third-party digital analysis software. The staining markers can be visualized at single-cell resolution, and cell phenotypes can be characterized via the co-localization of the markers (Figure 6 and Figure 7). The comprehensive analysis of a multiplexed image further reveals the tissue compartments, single-cell marker quantification, and nearest neighbor and proximity data (Figure 8).
A challenge in multiplexed image analysis is cell-type identification. Usually, when more single-object classifiers are applied to an image, more uncommon phenotypes will be annotated. Therefore, using known markers that are not co-expressed in the same classifier and applying only the phenotype-related classifier to the annotation of single cells are recommended. Variations in cell-type annotation will result in substantially different spatial results, such asdifferences in cell spatial distribution and cellular neighborhood analysis26,27.
Multiplexed image analysis has proven to be successful in staining and imaging many sample types, including FFPE tissue, fresh frozen tissue, archived whole slides, and tissue microarrays. Multiplexed images of breast, brain, lung, spleen, kidney, lymph node, and skin tissue sections can be acquired with deep single-cell spatial phenotyping data5,16,25,28.
In the future, more predesigned antibodiesfor multiplexed imaging are expected. Additionally, the development of specific software for multiplexed image analysis is greatly needed. Currently, many commercially available and open-source software programs for Hi-Plex image analysis exist29, but scientists still need help in creating a standard workflow for these analyses30,31. Although the composite images captured using this protocol are compatible with third-party software, this may result in extra costs for the user. Another disadvantage of the multiplexed imaging technology is the signal reduction in nuclear protein detection after iterative washing, hybridization, and stripping with large panels of antibodies. Fortunately, this can be minimized by retrieving the barcoded fluorophores at early cycles when designing the reporter plates. Recently, this platform was upgraded with a new high-speed scanning system, which has dramatically reduced the time to obtain composite images32. Additionally, a new strategy using tyramide-conjugated barcodes has been reported to enhance the oligonucleotide-conjugated antibody barcoding-based imaging. This technology aims to amplify staining signals for which barcode-conjugated antibodies are difficult to obtain33.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors thank Donald R. Norwood of Editing Services, Research Medical Library at MD Anderson for editing this article and the multiplex IF and image analysis laboratory in the Department of Translational Molecular Pathology at MD Anderson. This project was supported in part by the Translational Molecular Pathology-Immunoprofiling laboratory (TMP-IL) Moonshots Platform at the Department of Translational Molecular Pathology, The University of Texas MD Anderson Cancer Center and the NCI Cooperative Agreement U24CA224285 (to the MD Anderson Cancer Center CIMAC).
Materials
| 10x AR9 Buffer | Akoya Biosciences | AR900250ML | |
| 10x Buffer | Akoya Biosciences | 7000001 | |
| 16% paraformaldehyde | Thermo Fisher Scientific | 28906 | |
| 1X Antibody Diluent/Block | Akoya Biosciences | ARD1001EA | |
| Antibody Conjugation Kit | Akoya Biosciences | 7000009 | Contains Filter Blocking Solution, Antibody Reduction Solution 1, Antibody Reduction Solution 2, Conjugation Solution, Purification Solution, Antibody Storage Solution |
| Assay Reagent | Akoya Biosciences | 7000002 | |
| Diethyl pyrocarbonate | Sigma-Aldrich | 40718-25ML | |
| Dimethyl sulfoxide | Avantor/VWR | BDH1115-4LP | |
| Ethanol, 200 proof | |||
| G Blocker V2 | Akoya Biosciences | 240199 | |
| Histoclear | Thermo Fisher Scientific | 50-329-50 | |
| Methanol | Sigma-Aldrich | 34860-1L-R | |
| Milli-Q Integral 10 | Millipore | ZRXQ010WW | |
| Niknon Fluorescence microscope | Keyence Corp. of America | BZ-X810 | |
| Nuclear Stain | Akoya Biosciences | 7000003 | |
| Nuclease-free water | Thermo Fisher Scientific | AM9938 | |
| NuPAGE | Thermo Fisher Scientific | NP0008 | |
| PBS | Thermo Fisher Scientific | 14190136 | |
| PhenoCycler Barcodes/Reporters Combination | Akoya Biosciences | 5450004 (BX025/RX025) | |
| 5450003 (BX022/RX022) | |||
| 5450023 (BX002/RX002) | |||
| 5250002 (BX020/RX020) | |||
| 2520003 (BX023/RX023) | |||
| 5250005 (BX029/RX029) | |||
| 5250007 (BX035/RX035) | |||
| 5250012 (BX052/RX052) | |||
| 5550012 (BX030/RX030) | |||
| 5550015 (BX042/RX042) | |||
| 5550014 (BX036/RX036) | |||
| QuPath | Open-Source | https://qupath.github.io/ | |
| SimplyBlue SafeStain | Thermo Fisher Scientific | LC6065 | |
| Staining Kit | (Akoya Biosciences | 7000008 | Contains Hydration Buffer, N Blocker, J Blocker, S Blocker, Fixative Reagent, Storage Buffer |
References
- Hanahan, D., Coussens, L. M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell. 21 (3), 309-322 (2012).
- Labani-Motlagh, A., Ashja-Mahdavi, M., Loskog, A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune responses. Frontiers in Immunology. 11, 940 (2020).
- Jin, M. Z., Jin, W. L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduction and Targeted Therapy. 5 (1), 166 (2020).
- Waldman, A. D., Fritz, J. M., Lenardo, M. J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nature Reviews Immunology. 20 (11), 651-668 (2020).
- Black, S., et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nature Protocols. 16 (8), 3802-3835 (2021).
- Tan, W. C. C., et al. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Communications. 40 (4), 135-153 (2020).
- Radtke, A. J., et al. IBEX: A versatile multiplex optical imaging approach for deep phenotyping and spatial analysis of cells in complex tissues. Proceedings of the National Academy of Sciences of the United States of America. 117 (52), 33455-33465 (2020).
- Allam, M., Cai, S., Coskun, A. F. Multiplex bioimaging of single-cell spatial profiles for precision cancer diagnostics and therapeutics. NPJ Precision Oncology. 4, 11 (2020).
- Eng, J., et al. A framework for multiplex imaging optimization and reproducible analysis. Communications Biology. 5 (1), 438 (2022).
- Taube, J. M., et al. Multi-institutional TSA-amplified multiplexed immunofluorescence reproducibility evaluation (MITRE) study. Journal for Immunotherapy of Cancer. 9 (7), e002197 (2021).
- Binnewies, M., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine. 24 (5), 541-550 (2018).
- Heindl, A., Nawaz, S., Yuan, Y. Mapping spatial heterogeneity in the tumor microenvironment: A new era for digital pathology. Laboratory Investigation. 95 (4), 377-384 (2015).
- Kuswanto, W., Nolan, G., Lu, G. Highly multiplexed spatial profiling with CODEX: bioinformatic analysis and application in human disease. Seminars in Immunopathology. 45 (1), 145-157 (2022).
- Phillips, D., et al. Immune cell topography predicts response to PD-1 blockade in cutaneous T cell lymphoma. Nature Communications. 12 (1), 6726 (2021).
- Jhaveri, N., et al. Deep ultrahigh-plex spatial phenotyping of human cancer tissues. Cancer Research. 82, 3877 (2022).
- Goltsev, Y., et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. 174 (4), 968-981 (2018).
- Yuan, Y. Spatial heterogeneity in the tumor microenvironment. Cold Spring Harbor Perspectives in Medicine. 6 (8), a026583 (2016).
- Bruck, O., et al. Spatial immunoprofiling of the intratumoral and peritumoral tissue of renal cell carcinoma patients. Modern Pathology. 34 (12), 2229-2241 (2021).
- Tsujikawa, T., et al. Prognostic significance of spatial immune profiles in human solid cancers. Cancer Science. 111 (10), 3426-3434 (2020).
- Hoyt, C. C. Multiplex immunofluorescence and multispectral imaging: forming the basis of a clinical test platform for immuno-oncology. Frontiers in Molecular Biosciences. 8, 674747 (2021).
- Parra, E. R., et al. Identification of distinct immune landscapes using an automated nine-color multiplex immunofluorescence staining panel and image analysis in paraffin tumor tissues. Scientific Reports. 11 (1), 4530 (2021).
- Elaldi, R., et al. High dimensional imaging mass cytometry panel to visualize the tumor immune microenvironment contexture. Frontiers in Immunology. 12, 666233 (2021).
- Campos Clemente, L., Shi, O., Rojas, F., Parra, E. R. Expanding the comprehension of the tumor microenvironment using mass spectrometry imaging of formalin-fixed and paraffin-embedded tissue samples. Journal of Visualized Experiments. (184), e64015 (2022).
- Schurch, C. M., et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell. 182 (5), 1341-1359 (2020).
- Phillips, D., et al. Highly multiplexed phenotyping of immunoregulatory proteins in the tumor microenvironment by CODEX tissue imaging. Frontiers in Immunology. 12, 687673 (2021).
- Hickey, J. W., Tan, Y., Nolan, G. P., Goltsev, Y. Strategies for accurate cell type identification in CODEX multiplexed imaging data. Frontiers in Immunology. 12, 727626 (2021).
- Parra, E. R. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Frontiers in Molecular Biosciences. 8, 668340 (2021).
- Tzoras, E., et al. Dissecting tumor-immune microenvironment in breast cancer at a spatial and multiplex resolution. Cancers. 14 (8), 1999 (2022).
- Francisco-Cruz, A., Parra, E. R., Tetzlaff, M. T. Wistuba, II. Multiplex immunofluorescence assays. Methods in Molecular Biology. 2055, 467-495 (2020).
- Shakya, R., Nguyen, T. H., Waterhouse, N., Khanna, R. Immune contexture analysis in immuno-oncology: applications and challenges of multiplex fluorescent immunohistochemistry. Clinical and Translational Immunology. 9 (10), e1183 (2020).
- Wilson, C. M., et al. Challenges and opportunities in the statistical analysis of multiplex immunofluorescence data. Cancers. 13 (12), 3031 (2021).
- DeRosa, J. Setting a new standard for spatial omics: An integrated multiomics approach: every single cell, two different analytes, one unbiased picture. Genetic Engineering & Biotechnology News. 42, 26-28 (2022).
- Simonson, P. D., Valencia, I., Patel, S. S. Tyramide-conjugated DNA barcodes enable signal amplification for multiparametric CODEX imaging. Communications Biology. 5 (1), 627 (2022).
