Chiarire le basi molecolari delle funzioni cellulari spesso richiede l’espressione di DNA transgenico in coltura cellulare. Per essere espressi, i transgeni devono penetrare attraverso la membrana selettiva di una cellula e raggiungere il nucleo 1,2. Pertanto, la capacità di aggirare efficacemente le barriere fisiche della cellula e manipolare i suoi processi centrali è una necessità per applicare la transgenesi alla scoperta di nuovi fenomeni biologici. Un approccio sfrutta la capacità intrinseca dei virus di veicolare ed esprimere DNA estraneo 3,4.
Il virus adeno-associato (AAV) è uno dei più piccoli virus dei mammiferi: il suo genoma a DNA a singolo filamento di 4,7 kilobasi (kb) contiene due geni, rep (per la replicasi) e cap (per il capside), impacchettati all’interno di un capside icosaedrico di 60 mer che misura 25 nm. I geni rep/cap hanno più promotori, frame di lettura e prodotti di splicing che codificano per almeno nove proteine uniche necessarie per la replicazione, la produzione e il confezionamento virale 5,6. Inoltre, entrambe le estremità del genoma contengono strutture secondarie chiamate ripetizioni terminali invertite (ITR) che sono necessarie per la replicazione del DNA, l’impacchettamento del genoma e l’elaborazione a valle durante la trasduzione 7,8,9,10. Gli ITR sono gli unici elementi del DNA necessari per l’impacchettamento del genoma nel capside, e quindi l’AAV può essere clonato per scopi di trasporto del transgene sostituendo i geni rep/cap virali con elementi regolatori e/o geni di interesse6 scelti dal ricercatore. L’AAV ricombinante risultante (rAAV), con un genoma vettoriale ingegnerizzato (VG), è ampiamente utilizzato in clinica per la terapia genica umana e ha accumulato successi11. Un uso sottovalutato del vettore è in laboratorio; Gli rAAV possono raggiungere in modo efficiente l’espressione del transgene nelle cellule in coltura per soddisfare le esigenze sperimentali di un ricercatore12.
Il metodo più comune per la produzione di rAAV è la trasfezione a triplo plasmide in cellule HEK293 o 293T (Figura 1). Il primo plasmide, comunemente chiamato plasmide cis, contiene il transgene desiderato affiancato da ITR (pAAV). A seconda dell’applicazione, sono disponibili per l’acquisto plasmidi cis con elementi comuni, come promotori forti o strumenti basati su CRISPR. Il secondo è il plasmide pRep/Cap che contiene i geni wild-type AAV rep e cap forniti in-trans, cioè su un plasmide separato, non contenente ITR che esprime elementi regolatori e strutturali che poi interagiscono con il plasmide cis, ed è quindi chiamato plasmide trans. Oltre a racchiudere fisicamente il VG, il capside influenza il tropismo cellulare12,13. Fornendo il gene cap specifico per il sierotipo in-trans, i ricercatori sono facilmente in grado di massimizzare l’efficienza di trasduzione scegliendo un sierotipo del capside ottimizzato per la loro cellula bersaglio. Infine, come Dependoparvovirus, l’AAV richiede che un virus helper attivi l’espressione rep/cap dai suoi promotori virali, ottenuta dai geni helper adenovirali, forniti su un terzo plasmide come pAdΔF614,15. Dopo 72 ore di trasfezione a triplo plasmide, il vettore può essere rilasciato dalle cellule produttrici nel terreno di coltura mediante ripetuti cicli di congelamento/disgelo. L’intero contenuto della piastra viene quindi raccolto e i detriti cellulari di grandi dimensioni vengono rimossi mediante centrifugazione; il surnatante del terreno risultante è una preparazione grezza di rAAV pronta per le trasduzioni a valle.
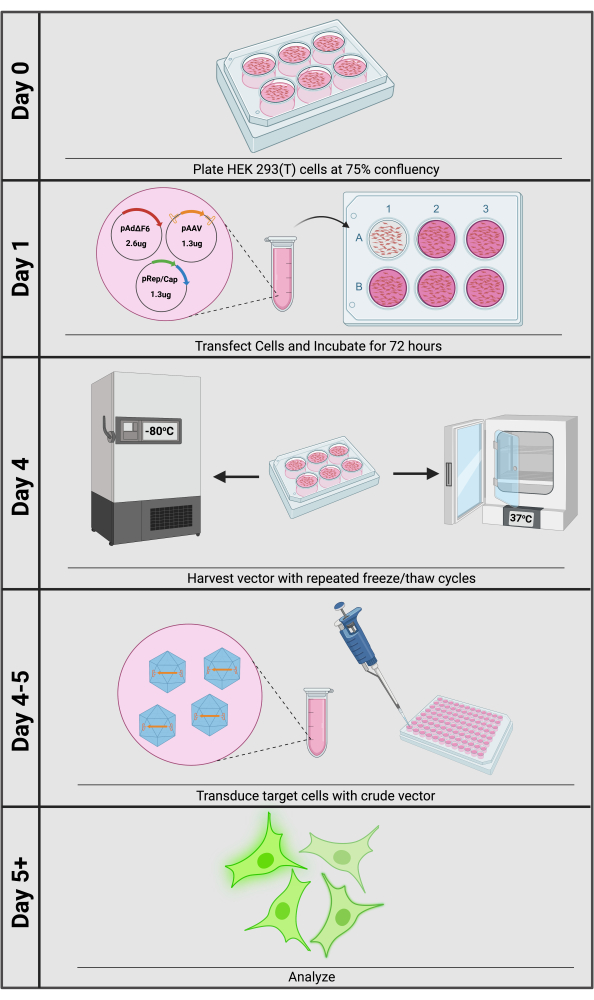
Figura 1: Panoramica della produzione di vettori rAAV grezzi. La produzione e la trasduzione di rAAV grezzi possono essere effettuate entro 5 giorni. Fare clic qui per visualizzare una versione più grande di questa figura.
rAAV può essere più favorevole per la somministrazione di transgeni rispetto ad altri metodi di trasfezione, che sono comunemente associati a tossicità cellulare, bassa efficienza e reagenti e attrezzature costosi, come per l’elettroporazione o la trasfezione a base chimica/lipidica16,17. rAAV aggira questi ostacoli e spesso fornisce una potente espressione transgenica con una tossicità minima e un tempo di intervento minimo. È importante sottolineare che la produzione di rAAV e la sua applicazione in coltura cellulare sono semplici e raramente richiedono la purificazione del vettore dal terreno di coltura (Figura 1). Inoltre, rAAV non integra il suo VG nel genoma dell’ospite, a differenza del rilascio del transgene lentivirale, e quindi riduce il rischio di mutagenesi inserzionale18. Nonostante i potenziali benefici dell’utilizzo di rAAV per la somministrazione di transgeni, è necessario considerare i limiti. È importante sottolineare che la dimensione del transgene, compresi gli ITR, non dovrebbe superare i 4,9 kb a causa dei vincoli fisici del capside, limitando così la capacità di un ricercatore di fornire efficacemente elementi regolatori e transgeni di grandi dimensioni. Inoltre, poiché rAAV è un virus non integrante, la trasduzione provoca un’espressione transgenica transitoria nelle cellule in divisione e potrebbe non essere pratica per un’espressione stabile. Tuttavia, i metodi che utilizzano il doppio Cas9 somministrato da rAAV e i modelli di riparazione diretta all’omologia (HDR) possono essere utilizzati per inserire stabilmente sequenze in loci genomici specifici, se un ricercatore lo desidera19.
Clonazione
Il protocollo di clonazione non è limitato al plasmide pAAV.CMV.Luc.IRES.EGFP.SV40 utilizzato in precedenza e può essere facilmente modificato in base alle esigenze sperimentali del ricercatore. Molti plasmidi contenenti ITR sono prontamente disponibili online per l’acquisto. Ad esempio, sono disponibili plasmidi contenenti sia Cas9 che un sito di clonaggio di sgRNA, ma richiedono alcuni passaggi aggiuntivi come la ricottura degli oligonucleotidi e il trattamento con PNK30. Inoltre, si possono trovare plasmidi contenenti un sito di clonazione multipla (MCS) con solo ITR e senza elementi regolatori interni31. Se devono essere utilizzati plasmidi diversi, gli enzimi di restrizione (RE) utilizzati per la digestione sono in genere gli unici elementi che potrebbero dover essere modificati in questo protocollo. Tuttavia, un limite dell’rAAV è la sua limitata capacità di carico. A causa delle limitazioni fisiche del capside, il genoma del vettore non dovrebbe superare i 4,9 kb, compresi gli ITR.
Quando si isola il plasmide dai batteri, è fondamentale utilizzare un kit midiprep o maxiprep a basso contenuto o privo di endotossine per mitigare i danni alle cellule durante la trasfezione o la trasduzione del triplo plasmide. I plasmidi dei kit miniprep contengono spesso impurità più elevate, concentrazioni ridotte e meno DNA superavvolto, che possono influenzare la produzione a valle di rAAV e quindi non sono raccomandati.
È fondamentale comprendere la struttura e le proprietà degli ITR durante la clonazione. In primo luogo, è estremamente difficile utilizzare la PCR attraverso l’ITR. I progetti di clonazione che richiedono l’amplificazione PCR tramite ITR dovrebbero essere evitati e dovrebbero inoltre limitare l’uso della tecnica di clonazione dell’assemblaggio Gibson. Pertanto, il clonaggio enzimatico di restrizione è il metodo preferito per la clonazione in plasmidi contenenti ITR. Inoltre, alcuni primer per il sequenziamento Sanger potrebbero non essere compatibili se la regione sequenziata contiene l’ITR. Invece, si consiglia di utilizzare primer che si allontanano dagli ITR e si inseriscono nel corpo del genoma del vettore per ottenere risultati di sequenziamento più precisi. In secondo luogo, gli ITR sono soggetti a delezioni, riarrangiamenti e mutazioni quando vengono trasformati in batteri per l’amplificazione plasmidica32,33. Per mitigare questi eventi, si consiglia di utilizzare ceppi batterici competenti carenti di ricombinazione, come Stbl3, e di incubarli a 30 °C per rallentare le divisioni cellulari. Infine, è stato osservato che le colonie più piccole possono corrispondere a cloni senza riarrangiamenti o delezioni, poiché quelle senza ITR possono conferire un vantaggio di crescita ed essere più grandi. Pertanto, si consiglia di scegliere colonie di piccole dimensioni.
Produzione vettoriale
Il successo della produzione del vettore rAAV può essere influenzato da più elementi. Un fattore critico è la salute delle cellule HEK293 o 293T utilizzate per la trasfezione. Generalmente, un basso numero di passaggi è l’ideale, poiché le cellule altamente passate possono presentare variazioni genotipiche e fenotipiche che possono ridurre i titoli di rAAV. Inoltre, la densità delle cellule seminate dovrebbe essere del 75%-90% di confluenza per una produzione efficace. Le cellule sparse generano basse rese vettoriali perché ci sono meno cellule disponibili per produrre vettori, mentre le cellule troppo cresciute non saranno trasfettate in modo efficiente.
Le variazioni tra i lotti di reagenti, le scorte di cellule e la variabilità generale da laboratorio a laboratorio contribuiscono alle differenze nell’efficienza di trasfezione e nel titolo di produzione. Un fattore ottimizzabile che può portare a miglioramenti del titolo è il rapporto plasmide:PEI nelle reazioni di trasfezione. È fondamentale utilizzare PEI MAX fresco (<1 mese). Si raccomanda di utilizzare un rapporto plasmide:PEI di 1:1 come punto di partenza e, se l'efficienza di trasfezione o trasduzione appare scarsa, testare diversi rapporti. L'ottimizzazione del titolo è più semplice se si utilizza un transgene con una lettura visiva, come il transgene reporter CMV.Luc.IRES.EGFP utilizzato in questo documento come materiale di partenza per la clonazione. Per eseguire l'ottimizzazione, seguire la fase 3 del protocollo utilizzando una piastra a 12 pozzetti e riducendo di due le masse del plasmide e i volumi dei reagenti (la massa finale del plasmide è di 2,6 μg). Regolare di conseguenza il volume PEI in modo che corrisponda a rapporti compresi tra 1:0,75 e 1:3, con incrementi crescenti di 0,25 (Figura 6). Diluire ogni reazione con 950 μL di terreno SF dopo 15 minuti. Per comodità, è possibile creare una miscela master contenente i plasmidi tripli e pipettare singolarmente in provette da 1,5 mL prima di aggiungere PEI (vedere il file supplementare 2). Raccogli il vettore, trasduci le cellule di interesse e l’immagine. Il pozzetto con la più alta efficienza di trasduzione (proporzione di cellule GFP+) corrisponde al titolo più alto e al rapporto ottimale di PEI:DNA.
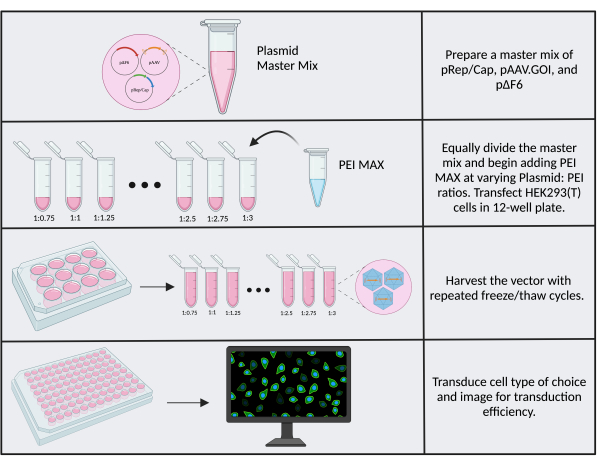
Figura 6: Flusso di lavoro di ottimizzazione PEI. Schema dei passaggi necessari per l’ottimizzazione PEI. Rapporti multipli di plasmidi: PEI vengono testati per determinare il rapporto ottimale. Fare clic qui per visualizzare una versione più grande di questa figura.
Considerazioni sul raccolto e sul titolo
La tecnica di congelamento/scongelamento utilizzata per raccogliere il vettore rAAV lisa efficacemente le cellule HEK293 in modo compatibile con l’uso diretto del lisato chiarificato per trasdurre le cellule in coltura. Alcuni sierotipi di rAAV, come AAV1, AAV8 e AAV9, vengono rilasciati dalle cellule durante la produzione del vettore e possono essere raccolti dal terreno cellulare coltivato senza cicli di congelamento/scongelamento34. Il metodo qui descritto produce in genere titoli dell’ordine di 1 x 1010 VG/mL quando si utilizzano capsidi AAV2 e 1 x10 11 VG/mL per AAV8. Sebbene titoli più elevati possano essere ottenuti mediante lisi detergente o altre sostanze chimiche, queste sono dannose per le cellule nell’uso a valle e richiedono che i rAAV siano ulteriormente purificati dal lisato. Il titolo più basso è un compromesso che un ricercatore dovrebbe considerare quando determina se i preparati grezzi sono appropriati per le proprie esigenze di ricerca, tuttavia, i titoli marginalmente più bassi prodotti dai metodi qui descritti possono trasdurre molto bene molti tipi di cellule (vedi risultati rappresentativi). Oltre all’efficienza di trasfezione e alla salute cellulare, i titoli dei vettori variano a seconda del capside utilizzato durante la produzione di rAAV e delle dimensioni e della sequenza del transgene all’interno del VG35.
Durante la raccolta di preparati vettoriali grezzi, può essere presente DNA plasmidico utilizzato durante la trasfezione a triplo plasmide e, sebbene raramente, provocare la trasfezione a valle durante la trasduzione. Inoltre, i VG non confezionati possono legarsi all’esterno dei capsidi e invocare una risposta immunitaria innata al DNA a singolo filamento nudo ed estraneo36,37. Pertanto, i tipi di cellule sensibili possono richiedere che le preparazioni vettoriali siano digerite e purificate con DNasi per rimuovere i VG e i plasmidi non confezionati.
Se si desidera calcolare il titolo di un preparato grezzo, è possibile eseguire la qPCR per quantificare il numero di VG confezionati all’interno di particelle resistenti alla DNasi (DRP). In breve, una piccola quantità di preparato grezzo viene digerita con DNasi per rimuovere il DNA plasmidico, contaminando gli acidi nucleici o il VG parzialmente confezionato. Il campione viene quindi sottoposto a qPCR e viene quantificato il VG protetto all’interno dei DRP, ottenendo un titolo con unità di genoma vettoriale per mL di preparato grezzo38. Non è consigliabile eseguire la titolazione vettoriale utilizzando saggi basati su ELISA che quantificano i titoli del capside. Rispetto al virus AAV wild-type, il rAAV soffre di una percentuale di capsidi vuoti e parzialmente impacchettati39. L’ELISA quantificherà tutti i capsidi indipendentemente dal loro contenuto genomico e sovrastimerà le unità trasducibili presenti in una preparazione, che richiede un VG impacchettato.
Considerazioni sulla trasduzione
Molti fattori influenzano le trasduzioni di rAAV e le opportune considerazioni dovrebbero essere fatte per ogni nuovo esperimento. A seconda del promotore che guida l’espressione del transgene, l’insorgenza dell’espressione può avvenire già 4 ore dopo la trasduzione (hpt) e il picco di espressione è tipicamente raggiunto entro 48 hpt. È importante tenere presente il periodo di tempo che intercorre tra la semina iniziale delle cellule e l’endpoint sperimentale. Questo per stimare la confluenza iniziale delle cellule e garantire che non crescano eccessivamente entro la fine dell’esperimento. Se le cellule diventano troppo confluenti, il comportamento cellulare può essere alterato a causa di una risposta allo stress e può confondere i risultati sperimentali. Alcuni tipi di cellule, come U2-OS, possono tollerare abbastanza bene la crescita eccessiva/inibizione da contatto. Inoltre, possono resistere a lunghi periodi (48 h+) in terreno condizionato privo di siero, il prodotto di questo protocollo di produzione. Tuttavia, i tipi di cellule sensibili possono richiedere l’aggiunta di siero o la diluizione del preparato grezzo con uno speciale terreno di crescita per mantenere la salute durante la trasduzione. Un’efficienza di trasduzione leggermente ridotta dall’uso di terreni contenenti siero è un potenziale compromesso per la salute delle cellule e dovrebbe essere presa in considerazione dal ricercatore.
Tipicamente, per celle che si dividono rapidamente, una confluenza iniziale di circa il 50% è ottimale per le applicazioni che saranno terminate a 48 hpt. Tuttavia, la confluenza può essere regolata di conseguenza in base alle esigenze dell’esperimento. Non è consigliabile trasdurre linee cellulari immortalizzate di tipo monostrato con una confluenza superiore al 75% a causa della diminuzione dell’efficienza di trasduzione. La maggior parte dei tipi di cellule in coltura viene trasdotta con successo e sana dopo l’incubazione notturna con preparati rAAV grezzi, seguita da un passaggio a terreni freschi contenenti siero al mattino.
Il sierotipo del capside è un fattore importante da considerare quando si produce rAAV per trasdurre una cellula bersaglio, poiché il capside è il determinante primario del tropismo cellulare e della successiva espressione del transgene13. AAV2 è un sierotipo ampiamente utilizzato grazie alla sua capacità di trasdurre efficacemente molti tipi di cellule in coltura12. Questa proprietà di AAV2 può essere attribuita ai proteoglicani dell’eparina solfato (HSPGs) che fungono da fattore di attacco primario per AAV2 e agli alti livelli di HSPGs sulle cellule in coltura dall’adattamento alla crescita in un piatto40. Altri capsidi, come AAV9, sono meno efficaci nel trasdurre i tipi di cellule larghe e possono essere spiegati dai loro fattori di attaccamento di dipendenza che non sono espressi in questo setting41. Pertanto, raccomandiamo AAV2 come capside di prima scelta nelle cellule in coltura se una cellula bersaglio desiderata non è stata precedentemente testata con rAAV in letteratura.
Si noti che una delle principali limitazioni dei preparati vettoriali grezzi è che sono inappropriati per la trasduzione di modelli animali. Gli studi in vivo richiedono che i preparati siano purificati e sottoposti a valutazione della qualità.
Considerazioni sull’espressione del transgene e sulla potenziale integrazione
Gli rAAV non determinano in modo affidabile l’espressione permanente del transgene. Nel corso del tempo, i VG possono essere silenziati e l’espressione transgenica può essere interrotta dopo diversi passaggi42. Inoltre, la maggior parte dei VG rimane episomiale e i rAAV non contengono le proteine Rep virali che medierebbero la frequente integrazione nel genoma dell’ospite come in un’infezione lisogenica virale wild-type o promuoverebbero la replicazione dei VG43. Di conseguenza, gli episomi nelle cellule trasdotte alla fine saranno diluiti tra le cellule figlie attraverso le divisioni.
L’integrazione a livello basale è una possibilità per tutto il materiale di DNA transgenico consegnato. Tuttavia, i VG contenenti ITR sono inclini all’integrazione a una frequenza più elevata44. Pertanto, l’espressione permanente di un transgene può essere osservata in un piccolo sottogruppo di cellule. Gli utenti dovrebbero considerare questa possibilità soprattutto quando utilizzano rAAV per fornire enzimi che tagliano il DNA, come Cas9, poiché le rotture a doppio filamento possono comportare una frequenza ancora maggiore di integrazione ed espressione permanente45. Sebbene ciò renda rAAV un buon candidato per la fornitura di modelli di riparazione diretti all’omologia per il tagging endogeno o l’aggiunta genica, la possibilità di inserimento di Cas9 dovrebbe essere considerata19,46.
