脂质是膜的主要成分,膜为细胞提供边界并实现细胞内区室化 1,2,3。脂质是两亲性的,具有极性头部基团和两个疏水性脂肪酸尾部;它们自组装成双层,以尽量减少疏水链与水的接触3,4。亲水性头部基团和疏水性尾部的各种组合导致生物膜中出现不同类别的脂质,例如甘油磷脂、鞘脂和甾醇(图 1)1,5,6。甘油磷脂是真核细胞膜的主要组成部分,由甘油磷酸盐、长链脂肪酸和低分子量7 的头基组成。脂质命名法基于头部基团的差异;实例包括磷脂酰胆碱(PC)、磷脂酰乙醇胺(PE)、磷脂酰丝氨酸(PS)、磷脂酰甘油(PG)、磷脂酰肌醇(PI)或未修饰的磷脂酸(PA)5,6。至于疏水尾部,长度和饱和度随骨架结构而变化。可能的组合很多,导致哺乳动物细胞中有数千种脂质物种6。膜脂质组成的变化导致不同的机械和结构膜特性,从而影响整合膜蛋白和外周蛋白的活性 2,6。
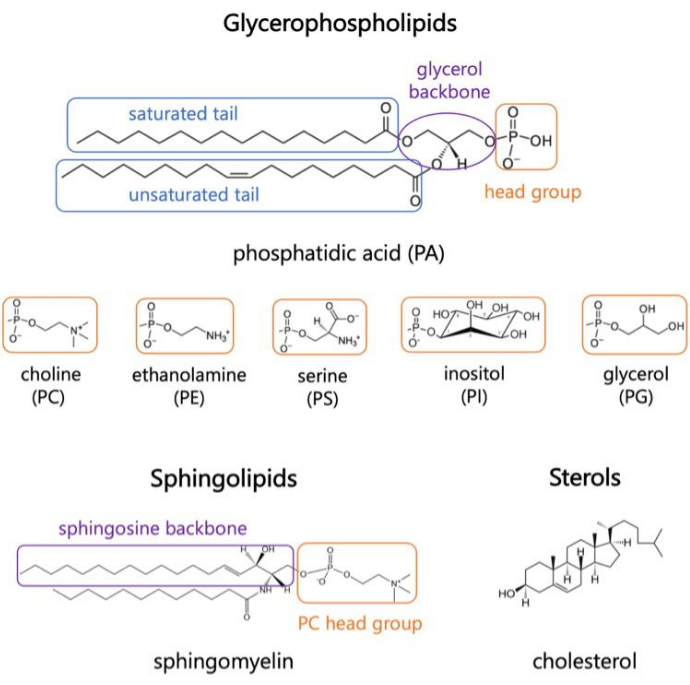
图 1.代表性脂质结构。 脂肪酸尾部以蓝色方框显示,常见脂质头部基团以橙色方框显示,样品骨架以紫色方框显示。 请点击这里查看此图的较大版本.
脂质是细胞过程、信号级联中的蛋白质激活和健康细胞稳态的活跃参与者 8,9。脂质动力学改变是感染的结果,也可能是疾病发病机制的标志物 10、11、12、13、14、15。作为细胞的屏障,膜脂质及其在小分子渗透中的作用的研究与药物递送系统和膜破坏机制有关16,17。细胞器、组织和生物体的化学多样性和脂质种类的不同比例产生了复杂的膜动力学2。因此,在脂质双层的建模研究中保留这些特性非常重要,特别是当研究的目标是检查其他生物分子与膜的相互作用时。在模型中要考虑的脂质种类取决于感兴趣的生物体和细胞区室。例如,PG 脂质对光合bateria 18 中的电子转移很重要,而磷酸化肌醇脂质 (PIP) 是哺乳动物细胞质膜 (PM) 动力学和信号级联的主要参与者19,20。在细胞内,PM、内质网 (ER)、高尔基体和线粒体膜含有影响其功能的独特脂质丰度。例如,内质网是脂质生物发生的枢纽,将胆固醇输送到PM和高尔基体;它含有高脂质多样性,富含 PC 和 PE,但甾醇含量低,可促进膜流动性21,22,23,24。相反,PM根据生物体25包含数百甚至数千种脂质,它含有高水平的鞘脂和胆固醇,与细胞24中的其他膜相比,使其具有特征性的刚性。对于像 PM 这样的膜,应考虑小叶不对称,PM的外小叶富含鞘磷脂、PC和胆固醇,而内小叶富含PE、PI和PS,这对信号级联反应很重要24。最后,脂质多样性还促进了在堆积和内部秩序上不同的微结构域的形成,称为脂筏24,26;它们表现出横向不对称性,据推测在细胞信号转导中起重要作用26,并且由于它们的瞬时性质而难以研究。
透视、光谱和模型膜系统(如巨型单层囊泡 (GUV))等实验技术已被用于研究生物分子与膜的相互作用。然而,仅靠实验方法很难捕捉到所涉及组件的复杂性和动态性。例如,在蛋白质跨膜结构域的成像、此类研究中使用的膜的复杂性以及感兴趣过程中中间或瞬态的鉴定方面存在局限性27,28,29。自 1980 年代脂质单层和双层分子模拟出现以来29,脂质-蛋白质系统及其相互作用现在可以在分子水平上进行量化。分子动力学 (MD) 模拟是一种常见的计算技术,它根据粒子的分子间作用力预测粒子的运动。加性相互作用电位描述了系统30的粒子之间的键合和非键合相互作用。用于模拟这些相互作用的参数集称为模拟力场 (FF)。这些参数是从从头开始计算、半经验和量子力学计算中获得的,并针对 X 射线和电子衍射实验、核磁共振、红外线、拉曼光谱和中子能谱等方法的再现数据进行了优化31.
MD仿真可用于研究分辨率为32,33,34的各种水平的系统。通过全原子 (AA) 模拟研究了旨在表征特定生物分子相互作用、氢键和其他高分辨率细节的系统。相比之下,粗粒度 (CG) 模拟将原子聚集成更大的官能团,以降低计算成本并检查更大规模的动力学33。介于这两者之间的是联合原子 (UA) 模拟,其中氢原子与它们各自的重原子结合以加速计算33,35。MD模拟是探索脂质膜动力学及其与其他分子相互作用的有力工具,可用于为膜界面感兴趣的过程提供分子水平机制。此外,MD模拟可用于缩小实验目标范围,并根据微观相互作用预测给定系统的大分子特性。
简而言之,给定一组初始坐标、速度和一组条件(如恒温和恒压),通过相互作用势和牛顿运动定律的数值积分来计算每个粒子的位置和速度。以迭代方式重复此操作,从而生成仿真轨迹30。这些计算是使用 MD 引擎执行的;在几个开源软件包中,GROMACS36 是最常用的引擎之一,也是我们在这里描述的引擎之一。它还包括用于分析和构建待模拟系统的初始坐标的工具37.其他 MD 发动机包括 NAMD38;CHARMM39 和 AMBER40,用户可以根据给定系统的计算性能自行选择。在仿真过程中可视化轨迹以及分析和解释结果至关重要。有多种工具可供选择;在这里,我们讨论了视觉分子动力学 (VMD),它提供了广泛的功能,包括具有扩展绘图和着色方法的三维 (3-D) 可视化、体积数据可视化、构建、准备和分析 MD 模拟系统的轨迹,以及对系统大小没有限制的轨迹电影制作,如果内存可用41,42,43。
系统组件之间预测动力学的准确性直接受到为轨迹传播选择的FF的影响。经验FF参数化工作由少数研究小组进行。MD 最成熟和最常见的 FF 包括 CHARMM39、AMBER 40、Martini44、OPLS 45 和 SIRAH 46。全原子加性CHARMM36(C36)力场47因其精确再现实验结构数据而广泛用于膜系统的AA MD。它最初由 CHARMM 社区开发,兼容 GROMACS 和 NAMD 等多个 MD 引擎。尽管在常见的FF中有所改进,但人们仍在不断努力改进参数集,以允许在对特定研究系统的兴趣的驱使下,密切再现实验可观察对象的预测48,49。
模拟脂质膜时的一个挑战是确定模拟轨迹的长度。这很大程度上取决于要分析的指标和旨在表征的过程。通常,复杂的脂质混合物需要更长的时间才能达到平衡,因为更多的物质必须有足够的时间在膜平面上扩散并达到稳定的横向组织。当感兴趣的属性达到一个平台并围绕一个常数值波动时,模拟被称为处于平衡状态。通常的做法是获得至少 100-200 ns 的平衡轨迹,以便对感兴趣的属性和相互作用进行适当的统计分析。通常在 200-500 ns 之间运行仅膜模拟,具体取决于脂质混合物的复杂性和研究问题。蛋白质-脂质相互作用通常需要更长的模拟时间,在 500-2000 ns 之间。使用膜系统加速采样和可观察动力学的一些方法是:(i)高度移动的膜模拟物(HMMM)模型,该模型用有机溶剂代替膜中脂质的末端碳以加速采样50;(ii)氢质量再分配(HMR),它将系统中重原子质量的一小部分与氢原子的质量结合起来,以允许使用更大的模拟时间步长51。
以下协议讨论了一种适合初学者的方法,用于使用 AA MD 构建、运行和分析真实的膜模型。鉴于 MD 模拟的性质,必须运行多个轨迹,以确保可重复性和对结果进行适当的统计分析。目前的做法是,每个感兴趣的系统至少运行三个副本。一旦为感兴趣的生物体和过程选择了脂质种类, 图2概述了构建、运行和分析纯膜系统模拟轨迹的基本步骤。
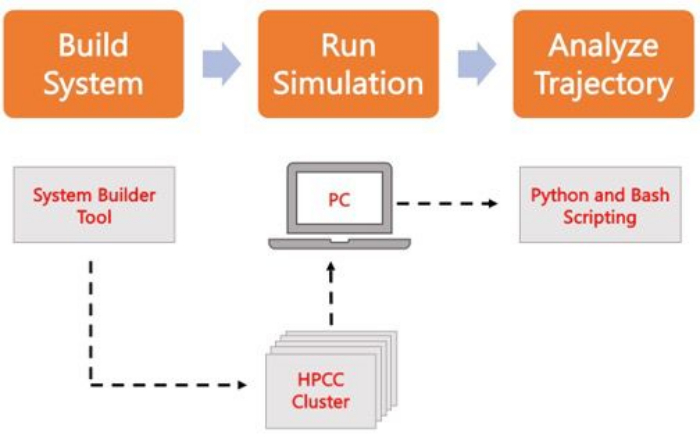
图2.运行 MD 仿真的原理图。 橙色框对应于协议中描述的三个主要步骤。下面是模拟过程的工作流程。在系统设置过程中,包含溶剂化膜系统初始坐标的系统是使用系统输入生成器(如 CHARMM-GUI Membrane Builder)构建的。将输入文件传输到高性能计算集群后,使用 MD 引擎(如 GROMACS)传播仿真轨迹。轨迹分析可以在计算机集群或本地工作站上进行,同时进行可视化。然后,使用带有内置分析代码(如 GROMACS 和 VMD)的包,或使用 Bash 脚本或各种 Python 库来执行分析。 请点击这里查看此图的较大版本.
