Lipider er vigtige bestanddele af membraner, som giver grænser for celler og muliggør intracellulær ruminddeling 1,2,3. Lipider er amfifile, med en polær hovedgruppe og to hydrofobe fedtsyrehaler; Disse samles selv i et dobbeltlag for at minimere kontakt mellem de hydrofobe kæder og vand 3,4. Forskellige kombinationer af hydrofile hovedgrupper og hydrofobe haler resulterer i forskellige klasser af lipider i biologiske membraner, såsom glycerophospholipider, sfingolipider og steroler (figur 1)1,5,6. Glycerophospholipider er primære byggesten i eukaryote cellemembraner sammensat af glycerophosphat, langkædede fedtsyrer og hovedgrupper med lav molekylvægt7. Lipidnomenklatur er baseret på forskelle i hovedgrupper; eksempler omfatter phosphatidylcholin (PC), phosphatidylethanolamin (PE), phosphatidylserin (PS), phosphatidylglycerol (PG), phosphatidyl-inositol (PI) eller umodificeret phosphatidsyre (PA)5,6. Hvad angår hydrofobe haler, varierer længden og graden af mætning sammen med rygradstrukturen. De mulige kombinationer er talrige, hvilket resulterer i tusindvis af lipidarter i pattedyrceller6. Ændringer i membranlipidsammensætningen fører til forskellige mekaniske og strukturelle membranegenskaber, der påvirker aktiviteten af både integrerede membranproteiner og perifere proteiner 2,6.
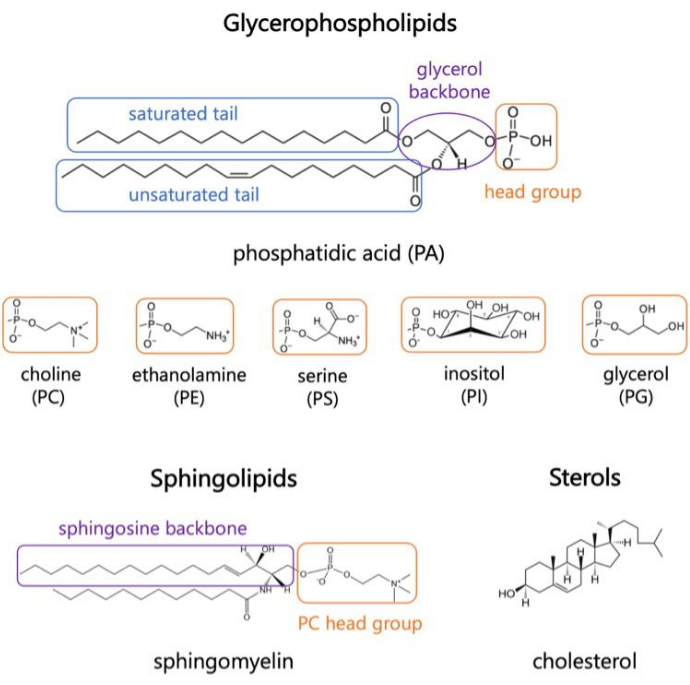
Figur 1. Repræsentative lipidstrukturer. Fedtsyrehaler vises i blå kasser, almindelige lipidhovedgrupper i orange og prøverygrad i lilla. Klik her for at se en større version af denne figur.
Lipider er aktive spillere i cellulære processer, proteinaktivering i signalkaskader og sund cellehomeostase 8,9. Ændret lipiddynamik er resultatet af infektion eller kan være markører for patogenese af sygdom 10,11,12,13,14,15. Som barrierer for cellen er undersøgelsen af membranlipider og deres rolle i gennemtrængning af små molekyler af relevans for lægemiddelafgivelsessystemer og membranforstyrrelsesmekanismer16,17. Kemisk mangfoldighed og forskellige forhold mellem lipidarter på tværs af organeller, væv og organismer giver anledning til kompleks membrandynamik2. Det er derfor vigtigt at bevare disse egenskaber i modelleringsstudier af lipiddobbeltlag, især når målet med en undersøgelse er at undersøge interaktioner mellem andre biomolekyler og membranen. De lipidarter, der skal overvejes i en model, afhænger af organismen og det cellulære rum af interesse. For eksempel er PG-lipider vigtige for elektronoverførsel i fotosyntetisk bateria18, mens phosphoryleret inositollipider (PIP’er) er vigtige aktører i plasmamembrandynamik (PM) og signalkaskader i pattedyrceller 19,20. Inde i cellen indeholder PM, endoplasmatisk retikulum (ER), Golgi og mitokondriemembraner unikke lipidmængder, der påvirker deres funktion. For eksempel er ER knudepunktet for lipidbiogenese og transporterer kolesterol ud til PM og Golgi; den indeholder en høj lipiddiversitet med en overflod af PC og PE, men lavt sterolindhold, hvilket fremmer membranfluiditet21,22,23,24. I modsætning hertil inkorporerer PM hundreder og endda tusinder af lipidarter afhængigt af organismen25, den indeholder høje niveauer af sphingolipider og kolesterol, der giver den en karakteristisk stivhed sammenlignet med andre membraner i cellen24. Brochureasymmetri bør overvejes for membraner som PM, som har en ydre folder rig på sphingomyelin, PC og kolesterol og en indre folder rig på PE, PI og PS, der er vigtige for signalering af kaskader24. Endelig beder lipiddiversitet også dannelsen af mikrodomæner, der adskiller sig i pakning og intern orden, kendt som lipidflåder24,26; Disse udviser lateral asymmetri, antages at spille vigtige roller i cellulær signalering26 og er svære at studere på grund af deres forbigående karakter.
Eksperimentelle teknikker såsom fluoroskopi, spektroskopi og modelmembransystemer som gigantiske unilamellære vesikler (GUV’er) er blevet brugt til at undersøge interaktioner mellem biomolekyler og membraner. Imidlertid er den komplekse og dynamiske karakter af de involverede komponenter vanskelig at fange med eksperimentelle metoder alene. For eksempel er der begrænsninger på billeddannelse af transmembrandomæner af proteiner, kompleksiteten af membraner, der anvendes i sådanne undersøgelser, og identifikation af mellemliggende eller forbigående tilstande under processen af interesse27,28,29. Siden fremkomsten af molekylær simulering af lipidmonolag og dobbeltlag i 1980’erne29 kan lipid-proteinsystemer og deres interaktioner nu kvantificeres på molekylært niveau. Simulering af molekylær dynamik (MD) er en almindelig beregningsteknik, der forudsiger partiklernes bevægelse baseret på deres intermolekylære kræfter. Et additivt interaktionspotentiale beskriver de bundne og ikke-bundne interaktioner mellem partikler i systemet30. Det sæt parametre, der bruges til at modellere disse interaktioner, kaldes simuleringskraftfeltet (FF). Disse parametre opnås fra ab initio-beregninger, semiempiriske og kvantemekaniske beregninger og optimeres til reproducerede data fra røntgen- og elektrondiffraktionseksperimenter, NMR, infrarød, Raman og neutronspektroskopi, blandt andre metoder31.
MD-simuleringer kan bruges til at studere systemer på forskellige niveauer af opløsning32,33,34. Systemer, der sigter mod at karakterisere specifikke biomolekylære interaktioner, hydrogenbindinger og andre detaljer med høj opløsning, studeres med all-atom (AA) simuleringer. I modsætning hertil klumper grovkornede (CG) simuleringer atomer i større funktionelle grupper for at reducere beregningsomkostningerne og undersøge dynamik i større skala33. Placeret imellem disse to er united-atom (UA) simuleringer, hvor hydrogenatomer kombineres med deres respektive tunge atomer for at fremskynde beregningen33,35. MD-simuleringer er et kraftfuldt værktøj til udforskning af dynamikken i lipidmembraner og deres interaktioner med andre molekyler og kan tjene til at tilvejebringe molekylære niveaumekanismer til processer af interesse ved membrangrænsefladen. Derudover kan MD-simuleringer tjene til at indsnævre eksperimentelle mål og forudsige makromolekylære egenskaber af et givet system baseret på mikroskopiske interaktioner.
Kort sagt, givet et sæt indledende koordinater, hastigheder og et sæt betingelser som konstant temperatur og tryk, beregnes positioner og hastigheder for hver partikel gennem numerisk integration af interaktionspotentialet og Newtons bevægelseslov. Dette gentages iterativt og genererer derved en simuleringsbane30. Disse beregninger udføres med en MD-motor; blandt flere open source-pakker er GROMACS36 en af de mest anvendte motorer og den, vi beskriver her. Det omfatter også værktøjer til analyse og konstruktion af indledende koordinater for systemer, der skal simuleres37. Andre MD-motorer omfatter NAMD38; CHARMM39 og AMBER40, som brugeren kan vælge efter eget skøn baseret på beregningsydelsen for et givet system. Det er afgørende at visualisere banerne under simuleringen samt til analyse og fortolkning af resultaterne. En række værktøjer er tilgængelige; her diskuterer vi visuel molekylær dynamik (VMD), der tilbyder en bred vifte af funktioner, herunder tredimensionel (3-D) visualisering med ekspansive tegnings- og farvelægningsmetoder, volumetrisk datavisualisering, opbygning, forberedelse og analyse af baner af MD-simuleringssystemer og banefilmfremstilling uden grænser for systemstørrelse, hvis hukommelsen er tilgængelig41,42,43.
Nøjagtigheden af forudsagt dynamik mellem systemkomponenter påvirkes direkte af FF valgt til udbredelse af banen. Empirisk FF-parametriseringsindsats forfølges af få forskningsgrupper. De mest etablerede og almindelige FF for MD omfatter CHARMM39, AMBER 40, Martini44, OPLS 45 og SIRAH 46. All-atom additiv CHARMM36 (C36) kraftfelt47 anvendes i vid udstrækning til AA MD af membransystemer, da det nøjagtigt gengiver eksperimentelle strukturelle data. Det blev oprindeligt udviklet af CHARMM-samfundet, og det er kompatibelt med flere MD-motorer som GROMACS og NAMD. På trods af forbedringer på tværs af almindelige FF’er er der en kontinuerlig indsats for at forbedre parametersættene for at muliggøre forudsigelser, der nøje gengiver eksperimentelle observerbare, drevet af interesser i bestemte undersøgelsessystemer48,49.
En udfordring ved simulering af lipidmembraner er at bestemme længden af simuleringsbanen. Dette afhænger i høj grad af de målinger, der skal analyseres, og den proces, man sigter mod at karakterisere. Typisk kræver komplekse lipidblandinger længere tid at nå ligevægt, da flere arter skal have tid nok til at diffundere på membranplanet og nå en stabil lateral organisation. En simulering siges at være i ligevægt, når egenskaben af interesse har nået et plateau og svinger omkring en konstant værdi. Det er almindelig praksis at opnå mindst 100-200 ns ligevægtsbane for at udføre passende statistisk analyse af egenskaber og interaktioner af interesse. Det er almindeligt at køre membransimuleringer mellem 200-500 ns, afhængigt af kompleksiteten af lipidblandingen og forskningsspørgsmålet. Protein-lipid-interaktioner kræver typisk længere simuleringstider, mellem 500-2000 ns. Nogle tilgange til at fremskynde prøveudtagning og observerbar dynamik med membransystemer er: (i) den meget mobile membranmimetiske (HMMM) model, som erstatter slutcarbonatomer af lipider i membranen med organisk opløsningsmiddel for at fremskynde prøveudtagning50; og ii) hydrogenmasseomfordeling (HMR), som kombinerer en brøkdel af masserne af tunge atomer i et system med masserne af hydrogenatomer for at muliggøre anvendelse af et større simuleringstidstrin51.
Følgende protokol diskuterer en begyndervenlig tilgang til at bygge, køre og analysere realistiske membranmodeller ved hjælp af AA MD. I betragtning af karakteren af MD-simuleringer skal der køres flere baner for at tage højde for reproducerbarhed og korrekt statistisk analyse af resultaterne. Det er almindelig praksis at køre mindst tre replikaer pr. interessesystem. Når lipidarterne er blevet udvalgt til organismen og processen af interesse, er grundlæggende trin til at opbygge, køre og analysere en simuleringsbane for et membran-eneste system skitseret og opsummeret i figur 2.
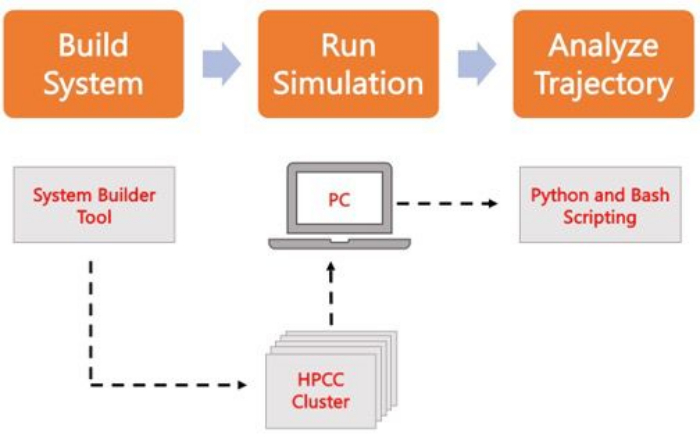
Figur 2. Skematisk til at køre MD-simuleringer. Orange bokse svarer til de tre hovedtrin, der er beskrevet i protokollen. Nedenunder er arbejdsgangen i simuleringsprocessen. Under systemopsætningen bygges systemet, der indeholder de indledende koordinater for et solvated membransystem, med en systemindgangsgenerator som CHARMM-GUI Membrane Builder. Efter overførsel af inputfilerne til en højtydende computerklynge formeres simuleringsbanen ved hjælp af en MD-motor, såsom GROMACS. Baneanalyse kan udføres på computerklyngen eller en lokal arbejdsstation sammen med visualisering. Analysen udføres derefter ved hjælp af enten pakker med indbygget analysekode såsom GROMACS og VMD eller ved hjælp af Bash-scripts eller forskellige Python-biblioteker. Klik her for at se en større version af denne figur.
