지질은 세포막의 주요 구성 요소로, 세포의 경계를 제공하고 세포 내 구획화를 가능하게 합니다 1,2,3. 지질은 양극성이며 극성 머리 그룹과 두 개의 소수성 지방산 꼬리가 있습니다. 이들은 소수성 사슬과 물 3,4의 접촉을 최소화하기 위해 이중층으로 자체 조립됩니다. 친수성 헤드 그룹과 소수성 꼬리의 다양한 조합은 생체막에서 글리세로인지질, 스핑고지질 및 스테롤과 같은 다양한 종류의 지질을 생성합니다(그림 1)1,5,6. 글리세로인지질(Glycerophospholipids)은 글리세로인산염(glycerophosphate), 장쇄 지방산(long-chain fatty acids), 저분자량의 머리(head) 그룹으로 구성된 진핵 세포막의 주요 구성 요소이다7. 지질 명명법은 머리 그룹의 차이를 기반으로 합니다. 예를 들면 포스파티딜콜린(PC), 포스파티딜-에탄올아민(PE), 포스파티딜세린(PS), 포스파티딜-글리세롤(PG), 포스파티딜-이노시톨(PI) 또는 변형되지 않은 포스파티드산(PA)5,6이 있습니다. 소수성 꼬리의 경우 포화도의 길이와 정도는 골격 구조와 함께 다양합니다. 가능한 조합은 셀 수 없이 많아 포유류 세포에서 수천 종의 지질이 생성된다6. 막 지질 조성의 변화는 통합 막 단백질과 말초 단백질 모두의 활성에 영향을 미치는 다양한 기계적 및 구조적 막 특성으로 이어집니다 2,6.
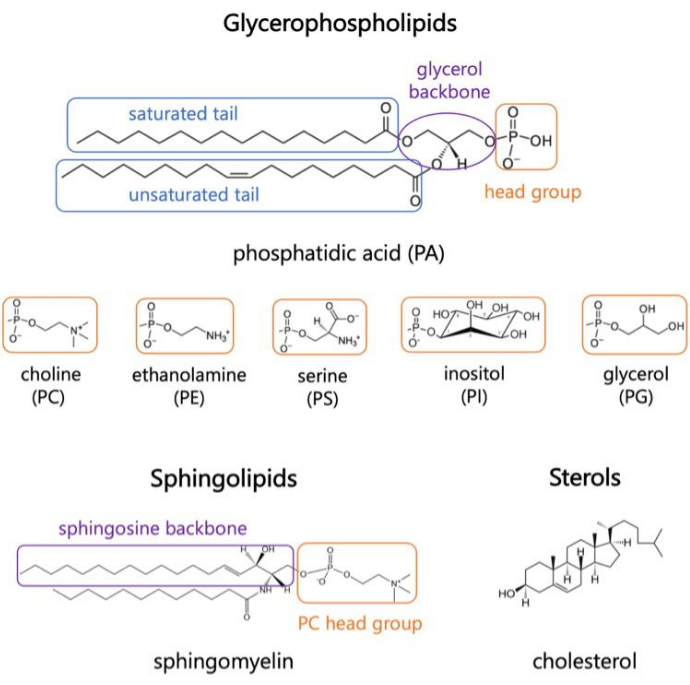
그림 1. 대표적인 지질 구조. 지방산 꼬리는 파란색 상자로, 일반적인 지질 머리 그룹은 주황색으로, 샘플 백본은 보라색으로 표시됩니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
지질은 세포 과정, 신호 전달 캐스케이드의 단백질 활성화, 건강한 세포 항상성 8,9에서 활발한 역할을 합니다. 변화된 지질 역학은 감염의 결과이거나 질병 10,11,12,13,14,15의 발병 기전의 표지자일 수 있다. 세포의 장벽으로서, 막 지질과 작은 분자의 투과에서의 역할에 대한 연구는 약물 전달 시스템 및 막 파괴 메커니즘과 관련이 있습니다16,17. 화학적 다양성과 세포기관, 조직 및 유기체에 걸친 지질 종의 다양한 비율은 복잡한 막 역학을 야기합니다2. 따라서 지질 이중층의 모델링 연구에서 이러한 특성을 유지하는 것이 중요하며, 특히 연구의 목표가 다른 생체 분자와 막의 상호 작용을 검사하는 것인 경우 더욱 그렇습니다. 모델에서 고려해야 할 지질 종은 관심 있는 유기체와 세포 구획에 따라 다릅니다. 예를 들어, PG 지질은 광합성 바테리아(18)에서 전자 전달에 중요한 반면, 인산화된 이노시톨 지질(PIP)은 포유류 세포에서 원형질막(PM) 역학 및 신호 캐스케이드의 주요 역할을 합니다19,20. 세포 내부의 PM, 소포체(ER), 골지체, 미토콘드리아 막에는 기능에 영향을 미치는 고유한 지질이 풍부하게 포함되어 있습니다. 예를 들어, 응급실은 지질 생물 발생의 허브이며 콜레스테롤을 PM과 골지로 운반합니다. PC와 PE가 풍부하고 지질 다양성이 높지만 스테롤 함량이 낮아 막 유동성21,22,23,24를 촉진합니다. 대조적으로, PM은 유기체(25)에 따라 수백, 심지어 수천 종의 지질 종을 포함하며, 세포(24)의 다른 막에 비해 특징적인 강성을 부여하는 높은 수준의 스핑고지질과 콜레스테롤을 함유하고 있다. 소엽 비대칭은 스핑고미엘린, PC, 콜레스테롤이 풍부한 외부 전단과 신호전달 캐스케이드에 중요한 PE, PI 및 PS가 풍부한 내부 전단이 있는 PM과 같은 막에 대해 고려해야 한다24. 마지막으로, 지질 다양성은 또한 지질 뗏목(lipid rafts)으로 알려진 패킹 및 내부 순서가 다른 미세 도메인(micro-domain)의 형성을 촉진한다(24,26); 이들은 측면 비대칭을 나타내고, 세포 신호전달에서 중요한 역할을 하는 것으로 추정되며(26), 일시적인 특성으로 인해 연구하기 어렵다.
형광투시, 분광법 및 거대 단층 소포(GUV)와 같은 모델 막 시스템과 같은 실험 기술은 생체 분자와 막의 상호 작용을 조사하는 데 사용되었습니다. 그러나 관련된 구성 요소의 복잡하고 역동적인 특성은 실험 방법만으로는 포착하기 어렵습니다. 예를 들어, 단백질의 막관통 도메인의 이미징, 이러한 연구에 사용되는 막의 복잡성 및 관심 과정 중 중간 또는 과도 상태의 식별에 제한이 있습니다27,28,29. 1980년대에 지질 단층과 이중층의 분자 시뮬레이션이 출현한 이후(29), 지질-단백질 시스템과 그 상호 작용은 이제 분자 수준에서 정량화될 수 있습니다. 분자 역학(MD) 시뮬레이션은 분자 간 힘을 기반으로 입자의 움직임을 예측하는 일반적인 계산 기술입니다. 부가적 상호작용 전위는 시스템(30)의 입자들 사이의 결합 및 비결합 상호작용을 설명한다. 이러한 상호 작용을 모델링하는 데 사용되는 파라미터 집합을 시뮬레이션 포스필드(FF)라고 합니다. 이러한 파라미터는 ab initio 계산, 반경험적 및 양자 역학적 계산으로부터 얻어지며, X선 및 전자 회절 실험, NMR, 적외선, 라만 및 중성자 분광법, 기타 방법(31)의 재현된 데이터에 최적화되어 있다.
MD 시뮬레이션은 해상도32,33,34의 다양한 수준에서 시스템을 연구하는 데 사용할 수 있습니다. 특정 생체 분자 상호 작용, 수소 결합 및 기타 고해상도 세부 사항을 특성화하는 것을 목표로 하는 시스템은 모든 원자(AA) 시뮬레이션으로 연구됩니다. 대조적으로, 거친 입자 (CG) 시뮬레이션은 계산 비용을 줄이고 더 큰 규모의 역학을 검사하기 위해 원자를 더 큰 작용기로 묶습니다33. 이 둘 사이에는 수소 원자가 각각의 무거운 원자와 결합하여 계산33,35을 가속화하는 단일 원자(UA) 시뮬레이션이 있습니다. MD 시뮬레이션은 지질막의 역학 및 다른 분자와의 상호 작용을 탐구하기 위한 강력한 도구이며 막 계면에서 관심 프로세스에 대한 분자 수준 메커니즘을 제공하는 역할을 할 수 있습니다. 또한 MD 시뮬레이션은 실험 대상의 범위를 좁히고 미세한 상호 작용을 기반으로 주어진 시스템의 고분자 특성을 예측하는 데 도움이 될 수 있습니다.
간단히 말해서, 초기 좌표, 속도 및 일정한 온도 및 압력과 같은 조건 집합이 주어지면 각 입자의 위치와 속도는 상호 작용 전위와 뉴턴의 운동 법칙의 수치적 적분을 통해 계산됩니다. 이것은 반복적으로 반복되어, 시뮬레이션 궤적(30)을 생성한다. 이러한 계산은 MD 엔진으로 수행됩니다. 여러 오픈 소스 패키지 중에서 GROMACS36은 가장 일반적으로 사용되는 엔진 중 하나이며 여기에서 설명하는 엔진입니다. 또한 시뮬레이션할 시스템의 초기 좌표를 분석하고 구성하기 위한 도구(37)를 포함한다. 다른 MD 엔진에는 NAMD38이 포함됩니다. CHARMM39 및 AMBER40은 사용자가 주어진 시스템의 계산 성능에 따라 재량에 따라 선택할 수 있습니다. 시뮬레이션 중에 궤적을 시각화하고 결과를 분석하고 해석하는 것이 중요합니다. 다양한 도구를 사용할 수 있습니다. 여기서는 광범위한 드로잉 및 채색 방법을 사용한 3차원(3차원) 시각화, 체적 데이터 시각화, MD 시뮬레이션 시스템의 궤적 구축, 준비 및 분석, 시스템 크기에 제한이 없는 궤적 동영상 제작(메모리를 사용할 수 있는 경우)을 포함하여 광범위한 기능을 제공하는 시각 분자 역학(VMD)에 대해 논의합니다(41,42,43).
시스템 구성 요소 간에 예측된 동역학의 정확도는 궤적의 전파를 위해 선택한 FF의 직접적인 영향을 받습니다. 경험적 FF 매개 변수화 노력은 소수의 연구 그룹에 의해 추구됩니다. MD에 대한 가장 확립되고 일반적인 FF에는 CHARMM39, AMBER 40, Martini44, OPLS 45 및 SIRAH 46이 있습니다. 모든 원자 첨가제 CHARMM36(C36) 역장(47)은 실험 구조 데이터를 정확하게 재현하기 때문에 멤브레인 시스템의 AA MD에 널리 사용됩니다. 원래 CHARMM 커뮤니티에서 개발했으며 GROMACS 및 NAMD와 같은 여러 MD 엔진과 호환됩니다. 공통 FF에 걸친 개선에도 불구하고, 특정 연구 시스템48,49에 대한 관심에 의해 주도된 실험적 관측 가능 물체를 밀접하게 재현하는 예측을 허용하기 위해 매개변수 세트를 개선하려는 지속적인 노력이 있습니다.
지질막을 시뮬레이션할 때 어려운 점은 시뮬레이션 궤적의 길이를 결정하는 것입니다. 이는 분석할 메트릭과 특성화하려는 프로세스에 따라 크게 달라집니다. 일반적으로 복잡한 지질 혼합물은 평형에 도달하는 데 더 오랜 시간이 필요한데, 이는 더 많은 종이 막 평면에서 확산되고 안정적인 측면 조직에 도달하는 데 충분한 시간이 필요하기 때문입니다. 시뮬레이션은 관심 속성이 안정기에 도달하고 일정한 값을 중심으로 변동할 때 평형 상태에 있다고 합니다. 관심 있는 특성 및 상호 작용에 대한 적절한 통계 분석을 수행하기 위해 최소 100-200ns의 평형 궤적을 얻는 것이 일반적입니다. 지질 혼합물의 복잡성과 연구 질문에 따라 200-500ns 사이의 멤브레인 전용 시뮬레이션을 실행하는 것이 일반적입니다. 단백질-지질 상호 작용은 일반적으로 500-2000ns 사이의 더 긴 시뮬레이션 시간이 필요합니다. 멤브레인 시스템으로 샘플링 및 관찰 가능한 동역학을 가속화하기 위한 몇 가지 접근법은 다음과 같다: (i) 멤브레인 내 지질의 말단 탄소를 유기 용매로 대체하여 샘플링을 가속화하는 HMMM(Highly Mobile Membrane Mimetic) 모델(50); 및 (ii) 수소 질량 재분할(HMR)은 시스템 내의 무거운 원자의 질량의 일부를 수소 원자의 질량과 결합하여 더 큰 시뮬레이션 시간단계(51)를 사용할 수 있도록 한다.
다음 프로토콜은 AA MD를 사용하여 사실적인 멤브레인 모델을 구축, 실행 및 분석하기 위한 초보자 친화적인 접근 방식에 대해 설명합니다. MD 시뮬레이션의 특성을 감안할 때 결과의 재현성과 적절한 통계 분석을 설명하기 위해 여러 궤적을 실행해야 합니다. 현재 관심 있는 시스템당 최소 3개의 복제본을 실행하는 것이 좋습니다. 관심 있는 유기체 및 공정에 대해 지질 종을 선택한 후에는 멤브레인 전용 시스템의 시뮬레이션 궤적을 구축, 실행 및 분석하기 위한 기본 단계가 그림 2에 요약되어 있습니다.
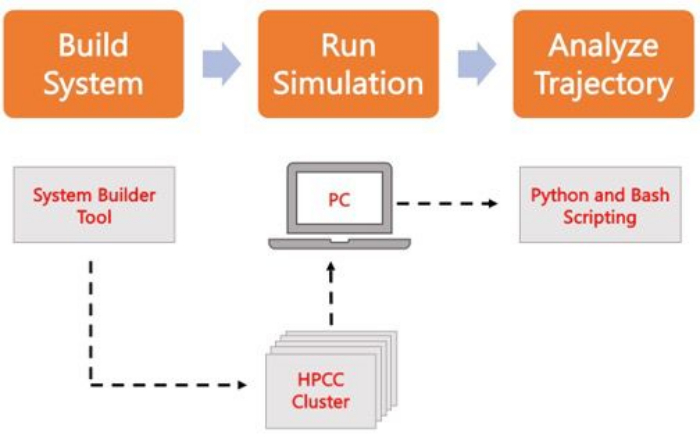
그림 2. MD 시뮬레이션을 실행하기 위한 회로도. 주황색 상자는 프로토콜에 설명된 세 가지 주요 단계에 해당합니다. 아래에는 시뮬레이션 프로세스의 작업 흐름이 있습니다. 시스템 설정 중에 용매 멤브레인 시스템의 초기 좌표를 포함하는 시스템은 CHARMM-GUI 멤브레인 빌더와 같은 시스템 입력 발생기로 구축됩니다. 입력 파일을 고성능 컴퓨팅 클러스터로 전송한 후 시뮬레이션 궤적은 GROMACS와 같은 MD 엔진을 사용하여 전파됩니다. 궤적 분석은 시각화와 함께 컴퓨터 클러스터 또는 로컬 작업 스테이션에서 수행할 수 있습니다. 그런 다음 GROMACS 및 VMD와 같은 분석 코드가 내장된 패키지를 사용하거나 Bash 스크립트 또는 다양한 Python 라이브러리를 사용하여 분석을 수행합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
