Липиды являются основными компонентами мембран, которые обеспечивают границы для клеток и обеспечивают внутриклеточную компартментализацию 1,2,3. Липиды амфифильные, с полярной головной группой и двумя гидрофобными хвостами жирных кислот; Они самоорганизуются в бислой, чтобы свести к минимуму контакт гидрофобных цепей с водой 3,4. Различные комбинации гидрофильных головных групп и гидрофобных хвостов приводят к образованию различных классов липидов в биологических мембранах, таких как глицерофосфолипиды, сфинголипиды и стерины (рис. 1)1,5,6. Глицерофосфолипиды являются первичными строительными блоками мембран эукариотических клеток, состоящих из глицерофосфата, длинноцепочечных жирных кислот и головных групп с низкой молекулярной массой7. Липидная номенклатура основана на различиях в головных группах; Например, фосфатидилхолин (ФК), фосфатидилэтаноламин (ПЭ), фосфатидилсерин (ФС), фосфатидилглицерин (ПГ), фосфатидил-инозитол (ПИ) или немодифицированная фосфатидная кислота (ПА)5,6. Что касается гидрофобных хвостов, то длина и степень насыщения варьируются, как и структура позвоночника. Возможные комбинации многочисленны, в результате чего в клетках млекопитающих образуются тысячи видов липидов6. Изменения липидного состава мембран приводят к различным механическим и структурным свойствам мембраны, которые влияют на активность как интегральных мембранных белков, так и периферических белков 2,6.
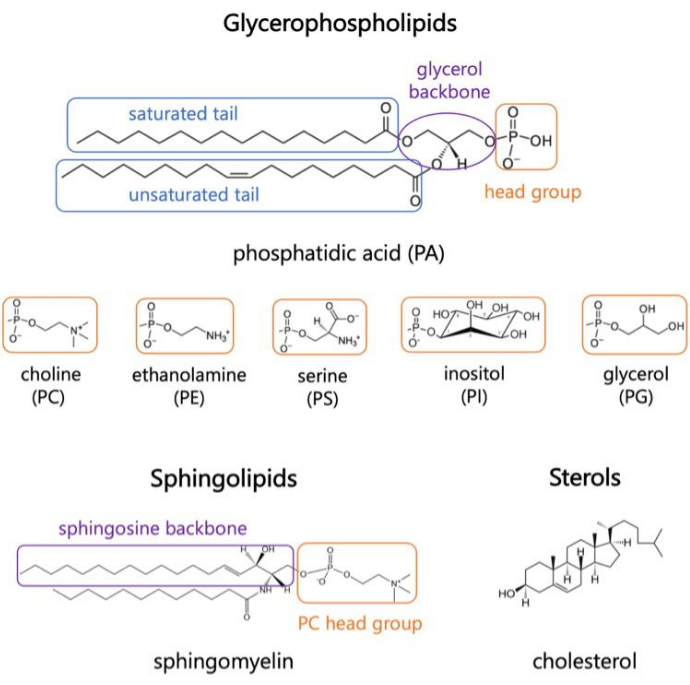
Рисунок 1. Репрезентативные липидные структуры. Хвосты жирных кислот показаны синими прямоугольниками, общие липидные группы головок — оранжевым, а костяк образцов — фиолетовым. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
Липиды являются активными игроками в клеточных процессах, активации белков в сигнальных каскадах и здоровом клеточном гомеостазе 8,9. Измененная липидная динамика является следствием инфекции или может быть маркерами патогенеза заболевания 10,11,12,13,14,15. Изучение мембранных липидов и их роли в проникновении малых молекул в качестве барьеров для клетки имеет значение для систем доставки лекарств и механизмов разрушения мембран16,17. Химическое разнообразие и различное соотношение видов липидов в органеллах, тканях и организмах приводят к сложной мембранной динамике. Поэтому важно сохранить эти характеристики при моделировании липидных бислоев, особенно когда целью исследования является изучение взаимодействия других биомолекул с мембраной. Виды липидов, которые следует учитывать в модели, зависят от организма и интересующего клеточного компартмента. Например, PG-липиды важны для переноса электронов в фотосинтетических bateria18, в то время как фосфорилированные инозитоловые липиды (PIPs) играют важную роль в динамике плазматической мембраны (PM) и сигнальных каскадах в клетках млекопитающих 19,20. Внутри клетки PM, эндоплазматический ретикулум (ER), мембраны Гольджи и митохондрии содержат уникальное количество липидов, которые влияют на их функцию. Например, ER является центром липидного биогенеза и транспортирует холестерин к PM и Гольджи; он содержит высокое липидное разнообразие с обилием ПК и ПЭ, но низкое содержание стеринов, что способствует текучести мембран21,22,23,24. Напротив, PM включает в себя сотни и даже тысячи видов липидов в зависимости от организма25, он содержит высокие уровни сфинголипидов и холестерина, которые придают ему характерную жесткость по сравнению с другими мембранами в клетке24. Асимметрию створок следует учитывать для мембран, таких как ПМ, которая имеет внешнюю створку, богатую сфингомиелином, ПК и холестерином, и внутреннюю створку, богатую ПЭ, ПИ и ФС, которые важны для сигнальных каскадов24. Наконец, липидное разнообразие также приводит к образованию микродоменов, различающихся по упаковке и внутреннему порядку, известных как липидные рафты24,26; Они демонстрируют латеральную асимметрию, предположительно играют важную роль в клеточнойсигнализации, и их трудно изучать из-за их переходной природы.
Экспериментальные методы, такие как рентгеноскопия, спектроскопия и модельные мембранные системы, такие как гигантские одноламеллярные везикулы (ГУВ), были использованы для исследования взаимодействия биомолекул с мембранами. Тем не менее, сложную и динамичную природу задействованных компонентов трудно охватить только экспериментальными методами. Например, существуют ограничения на визуализацию трансмембранных доменов белков, сложность мембран, используемых в таких исследованиях, и идентификацию промежуточных или переходных состояний во время интересующего процесса27,28,29. С момента появления молекулярного моделирования липидных монослоев и бислоев в 1980-х годах29 липидно-белковые системы и их взаимодействия теперь могут быть количественно оценены на молекулярном уровне. Моделирование молекулярной динамики (МД) — это распространенный вычислительный метод, который предсказывает движение частиц на основе их межмолекулярных сил. Потенциал аддитивного взаимодействия описывает связанные и несвязанные взаимодействия между частицами системы30. Набор параметров, используемых для моделирования этих взаимодействий, называется силовым полем моделирования (FF). Эти параметры получены в результате вычислений ab initio, полуэмпирических и квантово-механических расчетов и оптимизированы для воспроизведения данных рентгеновских и электронных дифракционных экспериментов, ЯМР, инфракрасной, комбинационной и нейтронной спектроскопии, а также других методов31.
МД-моделирование может быть использовано для исследования систем с различными уровнями разрешения32,33,34. Системы, целью которых является характеристика конкретных биомолекулярных взаимодействий, водородных связей и других деталей с высоким разрешением, изучаются с помощью моделирования всех атомов (АА). В отличие от этого, крупнозернистое (CG) моделирование объединяет атомы в более крупные функциональные группы для снижения вычислительных затрат иизучения крупномасштабной динамики. Между этими двумя моделями находятся модели объединенных атомов (UA), в которых атомы водорода объединяются с соответствующими тяжелыми атомами для ускорения вычислений33,35. Моделирование МД является мощным инструментом для изучения динамики липидных мембран и их взаимодействия с другими молекулами и может служить для создания механизмов на молекулярном уровне для интересующих процессов на границе раздела мембран. Кроме того, МД-моделирование может служить для сужения экспериментальных мишеней и прогнозирования макромолекулярных свойств данной системы на основе микроскопических взаимодействий.
Короче говоря, имея набор начальных координат, скоростей и набор условий, таких как постоянная температура и давление, положения и скорости каждой частицы вычисляются путем численного интегрирования потенциала взаимодействия и закона движения Ньютона. Это повторяется итеративно, тем самым генерируя траекториюмоделирования 30. Эти вычисления выполняются с помощью движка MD; Среди нескольких пакетов с открытым исходным кодом GROMACS36 является одним из наиболее часто используемых движков, и именно его мы опишем здесь. Он также включает в себя инструменты для анализа и построения начальных координат моделируемых систем37. Другие двигатели MD включают NAMD38; CHARMM39 и AMBER40, которые пользователь может выбрать по своему усмотрению в зависимости от вычислительной производительности данной системы. Очень важно визуализировать траектории во время моделирования, а также для анализа и интерпретации результатов. Доступны различные инструменты; Здесь мы обсудим визуальную молекулярную динамику (VMD), которая предлагает широкий спектр возможностей, включая трехмерную (3-D) визуализацию с расширенными методами рисования и раскрашивания, визуализацию объемных данных, построение, подготовку и анализ траекторий систем моделирования МД, а также создание траекторных фильмов без ограничений по размеру системы, если доступна память41,42,43.
На точность предсказанной динамики между компонентами системы напрямую влияет ФФ, выбранный для распространения траектории. Эмпирическая параметризация ФФ предпринимается несколькими исследовательскими группами. Наиболее распространенными и распространенными FF для MD являются CHARMM39, AMBER 40, Martini44, OPLS 45 и SIRAH 46. Силовое поле47 из всех атомов CHARMM36 (C36) широко используется для АА МД мембранных систем, так как оно точно воспроизводит экспериментальные структурные данные. Первоначально он был разработан сообществом CHARMM и совместим с несколькими движками MD, такими как GROMACS и NAMD. Несмотря на усовершенствования во всех распространенных FF, предпринимаются постоянные усилия по совершенствованию наборов параметров, позволяющих делать прогнозы, точно воспроизводящие экспериментальные наблюдаемые объекты, что обусловлено интересами к конкретным системам исследования48,49.
Сложность при моделировании липидных мембран заключается в определении длины траектории моделирования. Это во многом зависит от анализируемых показателей и процесса, который мы хотим охарактеризовать. Как правило, сложным липидным смесям требуется больше времени для достижения равновесия, так как большее количество видов должно иметь достаточно времени, чтобы диффундировать в плоскости мембраны и достичь стабильной латеральной организации. Моделирование называется равновесным, когда интересующее свойство достигло плато и колеблется около постоянной величины. Общепринятой практикой является получение по крайней мере 100-200 нс равновесной траектории для проведения соответствующего статистического анализа интересующих свойств и взаимодействий. Обычно моделирование только мембраны выполняется в диапазоне от 200 до 500 нс, в зависимости от сложности липидной смеси и исследовательского вопроса. Белок-липидные взаимодействия обычно требуют более длительного времени моделирования, от 500 до 2000 нс. Некоторые подходы к ускорению отбора проб и наблюдаемой динамике с помощью мембранных систем включают: (i) высокоподвижную мембранно-миметическую модель (HMMM), в которой конечные углероды липидов в мембране заменяются органическим растворителем для ускорения отбора проб50; и (ii) перераспределение массы водорода (HMR), которое объединяет часть масс тяжелых атомов в системе с массами атомов водорода, что позволяет использовать больший временной шаг51 моделирования.
В следующем протоколе описывается удобный для начинающих подход к построению, запуску и анализу реалистичных мембранных моделей с использованием AA MD. Учитывая природу моделирования МД, необходимо использовать несколько траекторий для обеспечения воспроизводимости и надлежащего статистического анализа результатов. В настоящее время рекомендуется запускать не менее трех реплик на интересующую систему. После того, как липидные формы были выбраны для интересующего организма и процесса, основные шаги по построению, запуску и анализу траектории моделирования мембранной системы описаны и обобщены на рисунке 2.
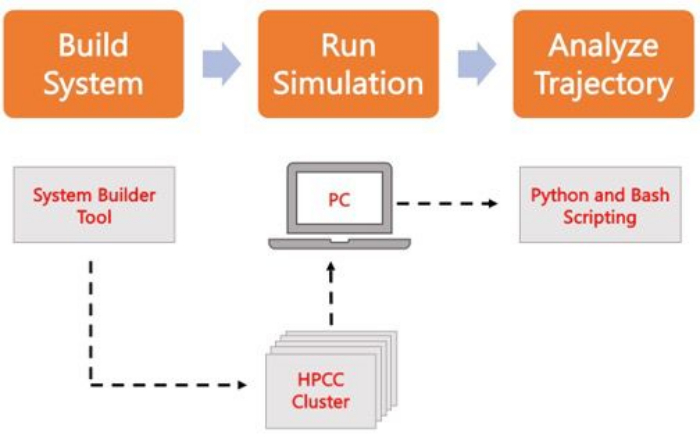
Рисунок 2. Schematic для запуска MD-моделирования. Оранжевые прямоугольники соответствуют трем основным шагам, описанным в протоколе. Ниже показан рабочий процесс моделирования. Во время настройки системы система, содержащая начальные координаты сольватированной мембранной системы, строится с помощью системного генератора входных данных, такого как CHARMM-GUI Membrane Builder. После передачи входных файлов в кластер высокопроизводительных вычислений траектория моделирования распространяется с помощью движка MD, такого как GROMACS. Анализ траектории может быть выполнен на вычислительном кластере или локальной рабочей станции вместе с визуализацией. Затем анализ выполняется либо с помощью пакетов со встроенным кодом анализа, таких как GROMACS и VMD, либо с помощью сценариев Bash или различных библиотек Python. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
