Lipide sind Hauptbestandteile von Membranen, die den Zellen Grenzen bilden und eine intrazelluläre Kompartimentierung ermöglichen 1,2,3. Lipide sind amphiphil, mit einer polaren Kopfgruppe und zwei hydrophoben Fettsäureschwänzen; Diese fügen sich selbst zu einer Doppelschicht zusammen, um den Kontakt der hydrophoben Ketten mit Wasser zu minimieren 3,4. Verschiedene Kombinationen von hydrophilen Kopfgruppen und hydrophoben Schwänzen führen zu unterschiedlichen Klassen von Lipiden in biologischen Membranen, wie z. B. Glycerophospholipide, Sphingolipide und Sterole (Abbildung 1)1,5,6. Glycerophospholipide sind primäre Bausteine eukaryotischer Zellmembranen, die aus Glycerophosphat, langkettigen Fettsäuren und Kopfgruppen mit niedrigem Molekulargewichtbestehen 7. Die Lipidnomenklatur basiert auf Unterschieden in den Kopfgruppen; Beispiele hierfür sind Phosphatidylcholin (PC), Phosphatidyl-Ethanolamin (PE), Phosphatidyl-Serin (PS), Phosphatidyl-Glycerin (PG), Phosphatidyl-Inositol (PI) oder die unmodifizierte Phosphatidsäure (PA)5,6. Bei hydrophoben Schwänzen variieren die Länge und der Sättigungsgrad sowie die Struktur des Rückgrats. Die möglichen Kombinationen sind zahlreich, was zu Tausenden von Lipidspezies in Säugetierzellen führt6. Veränderungen in der Zusammensetzung der Membranlipide führen zu unterschiedlichen mechanischen und strukturellen Membraneigenschaften, die sich auf die Aktivität sowohl der integralen Membranproteine als auch der peripheren Proteine auswirken 2,6.
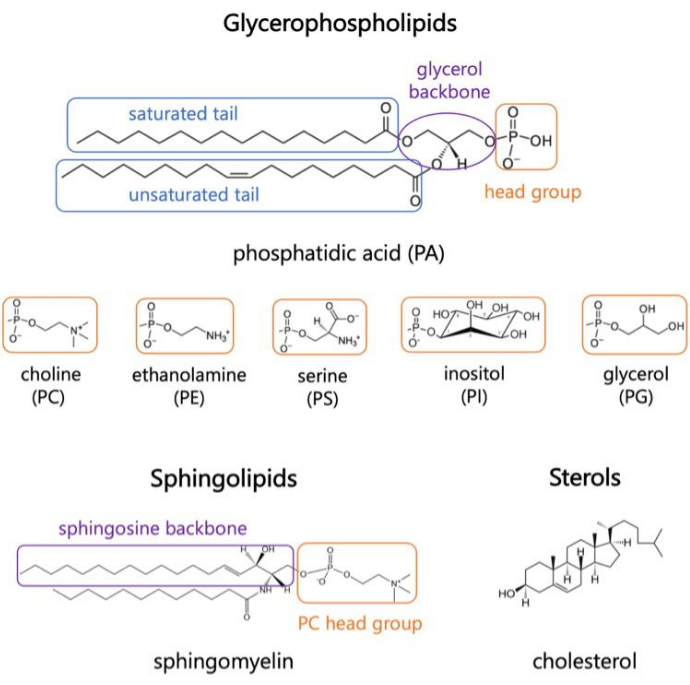
Abbildung 1. Repräsentative Lipidstrukturen. Fettsäureschwänze sind in blauen Kästchen, häufige Lipidkopfgruppen in Orange und Probenrückgrate in Lila dargestellt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Lipide sind aktive Akteure in zellulären Prozessen, Proteinaktivierung in Signalkaskaden und gesunder Zellhomöostase 8,9. Eine veränderte Lipiddynamik ist die Folge einer Infektion oder kann Marker für die Pathogenese der Erkrankung sein 10,11,12,13,14,15. Als Barrieren für die Zelle ist die Untersuchung von Membranlipiden und ihrer Rolle bei der Permeation kleiner Moleküle von Bedeutung für Arzneimittelverabreichungssysteme und Membranaufschlussmechanismen 16,17. Chemische Vielfalt und unterschiedliche Verhältnisse von Lipidspezies in Organellen, Geweben und Organismen führen zu einer komplexen Membrandynamik2. Daher ist es wichtig, diese Eigenschaften bei Modellierungsstudien von Lipiddoppelschichten beizubehalten, insbesondere wenn das Ziel einer Studie darin besteht, Wechselwirkungen anderer Biomoleküle mit der Membran zu untersuchen. Welche Lipidspezies in einem Modell zu berücksichtigen sind, hängt vom Organismus und dem Zellkompartiment ab, das von Interesse ist. Zum Beispiel sind PG-Lipide wichtig für den Elektronentransfer in photosynthetischen Baterien18, während phosphorylierte Inositol-Lipide (PIPs) wichtige Akteure bei der Dynamik der Plasmamembran (PM) und den Signalkaskaden in Säugetierzellen sind19,20. Im Inneren der Zelle enthalten das PM, das endoplasmatische Retikulum (ER), der Golgi und die mitochondrialen Membranen einzigartige Lipidhäufigkeiten, die ihre Funktion beeinflussen. Zum Beispiel ist das ER die Drehscheibe für die Lipidbiogenese und transportiert Cholesterin zu PM und Golgi; Es enthält eine hohe Lipidvielfalt mit einer Fülle von PC und PE, aber einen niedrigen Sterolgehalt, der die Membranfluidität fördert21,22,23,24. Im Gegensatz dazu enthält die PM je nach Organismus Hunderte und sogar Tausende von Lipidspezies25, sie enthält hohe Konzentrationen von Sphingolipiden und Cholesterin, die ihr eine charakteristische Steifigkeit im Vergleich zu anderen Membranen in der Zelle verleihen24. Die Asymmetrie des Segels sollte für Membranen wie die PM in Betracht gezogen werden, die ein äußeres Segel hat, das reich an Sphingomyelin, PC und Cholesterin ist, und ein inneres Segel, das reich an PE, PI und PS ist, die für Signalkaskaden wichtig sind24. Schließlich führt die Lipidvielfalt auch zur Bildung von Mikrodomänen, die sich in Packung und innerer Ordnung unterscheiden, die als Lipid-Rafts bekanntsind 24,26; Diese weisen eine laterale Asymmetrie auf, es wird angenommen, dass sie eine wichtige Rolle bei der zellulären Signalübertragungspielen 26, und sind aufgrund ihrer vorübergehenden Natur schwer zu untersuchen.
Experimentelle Techniken wie Fluoroskopie, Spektroskopie und Modellmembransysteme wie riesige unilamellare Vesikel (GUVs) wurden verwendet, um Wechselwirkungen von Biomolekülen mit Membranen zu untersuchen. Die Komplexität und Dynamik der beteiligten Komponenten lässt sich jedoch nur schwer mit experimentellen Methoden erfassen. Zum Beispiel gibt es Einschränkungen bei der Abbildung von Transmembrandomänen von Proteinen, der Komplexität von Membranen, die in solchen Studien verwendet werden, und der Identifizierung von intermediären oder vorübergehenden Zuständen während des interessierenden Prozesses27,28,29. Seit dem Aufkommen der molekularen Simulation von Lipid-Mono- und Doppelschichten in den 1980er Jahren29 können Lipid-Protein-Systeme und ihre Wechselwirkungen auf molekularer Ebene quantifiziert werden. Die Molekulardynamik-Simulation (MD) ist eine gängige Berechnungstechnik, die die Bewegung von Partikeln auf der Grundlage ihrer intermolekularen Kräfte vorhersagt. Ein additives Wechselwirkungspotential beschreibt die gebundenen und nicht gebundenen Wechselwirkungen zwischen Teilchen des Systems30. Der Satz von Parametern, die zur Modellierung dieser Wechselwirkungen verwendet werden, wird als Simulationskraftfeld (FF) bezeichnet. Diese Parameter werden aus ab initio-Berechnungen, semi-empirischen und quantenmechanischen Berechnungen gewonnen und optimiert, um unter anderem Daten aus Röntgen- und Elektronenbeugungsexperimenten, NMR-, Infrarot-, Raman- und Neutronenspektroskopie zu reproduzieren31.
MD-Simulationen können verwendet werden, um Systeme mit verschiedenen Auflösungsstufenzu untersuchen 32,33,34. Systeme, die darauf abzielen, spezifische biomolekulare Wechselwirkungen, Wasserstoffbrückenbindungen und andere hochauflösende Details zu charakterisieren, werden mit All-Atom-Simulationen (AA) untersucht. Im Gegensatz dazu werden bei grobkörnigen (CG) Simulationen Atome in größere funktionelle Gruppen zusammengefasst, um den Rechenaufwand zu senken und die Dynamik auf größerer Skala zu untersuchen33. Dazwischen befinden sich United-Atom (UA)-Simulationen, bei denen Wasserstoffatome mit ihren jeweiligen schweren Atomen kombiniert werden, um die Berechnung zu beschleunigen33,35. MD-Simulationen sind ein leistungsfähiges Werkzeug zur Erforschung der Dynamik von Lipidmembranen und ihrer Wechselwirkungen mit anderen Molekülen und können dazu dienen, Mechanismen auf molekularer Ebene für Prozesse von Interesse an der Membranschnittstelle bereitzustellen. Darüber hinaus können MD-Simulationen dazu dienen, experimentelle Ziele einzugrenzen und makromolekulare Eigenschaften eines bestimmten Systems auf der Grundlage mikroskopischer Wechselwirkungen vorherzusagen.
Kurz gesagt, bei einer Reihe von Anfangskoordinaten, Geschwindigkeiten und einer Reihe von Bedingungen wie konstanter Temperatur und konstantem Druck werden Positionen und Geschwindigkeiten jedes Teilchens durch numerische Integration des Wechselwirkungspotentials und des Newtonschen Bewegungsgesetzes berechnet. Dies wird iterativ wiederholt, wodurch eine Simulationstrajektorie30 erzeugt wird. Diese Berechnungen werden mit einer MD-Engine durchgeführt. Unter mehreren Open-Source-Paketen ist GROMACS36 eine der am häufigsten verwendeten Engines und diejenige, die wir hier beschreiben. Es enthält auch Werkzeuge für die Analyse und Konstruktion der Anfangskoordinaten der zu simulierenden Systeme37. Weitere MD-Motoren sind NAMD38; CHARMM39 und AMBER40, die der Benutzer nach eigenem Ermessen basierend auf der Rechenleistung eines bestimmten Systems auswählen kann. Es ist wichtig, die Trajektorien während der Simulation sowie für die Analyse und Interpretation der Ergebnisse zu visualisieren. Es steht eine Vielzahl von Werkzeugen zur Verfügung; Hier diskutieren wir die visuelle Molekulardynamik (VMD), die eine breite Palette von Funktionen bietet, einschließlich dreidimensionaler (3D) Visualisierung mit umfangreichen Zeichen- und Färbemethoden, volumetrischer Datenvisualisierung, Erstellen, Vorbereiten und Analysieren von Trajektorien von MD-Simulationssystemen und Erstellen von Trajektorienfilmen ohne Begrenzung der Systemgröße, wenn der Speicher verfügbar ist41,42,43.
Die Genauigkeit der vorhergesagten Dynamik zwischen den Systemkomponenten wird direkt durch die FF beeinflusst, die für die Ausbreitung der Trajektorie gewählt wird. Empirische FF-Parametrisierungsbemühungen werden von wenigen Forschungsgruppen verfolgt. Zu den etabliertesten und gebräuchlichsten FF für MD gehören CHARMM39, AMBER 40, Martini44, OPLS 45 und SIRAH 46. Das Vollatom-Additiv CHARMM36 (C36) Kraftfeld47 wird häufig für AA MD von Membransystemen verwendet, da es experimentelle Strukturdaten genau reproduziert. Es wurde ursprünglich von der CHARMM-Community entwickelt und ist mit mehreren MD-Engines wie GROMACS und NAMD kompatibel. Trotz Verbesserungen in den gängigen FFs gibt es kontinuierliche Bemühungen, die Parametersätze zu verbessern, um Vorhersagen zu ermöglichen, die experimentelle Observablen genau reproduzieren, angetrieben von Interessen an bestimmten Studiensystemen48,49.
Eine Herausforderung bei der Simulation von Lipidmembranen ist die Bestimmung der Länge der Simulationstrajektorie. Dies hängt weitgehend von den zu analysierenden Metriken und dem Prozess ab, den man charakterisieren möchte. Typischerweise benötigen komplexe Lipidmischungen mehr Zeit, um ein Gleichgewicht zu erreichen, da mehr Spezies genügend Zeit haben müssen, um auf der Membranebene zu diffundieren und eine stabile laterale Organisation zu erreichen. Eine Simulation befindet sich im Gleichgewicht, wenn die interessierende Eigenschaft ein Plateau erreicht hat und um einen konstanten Wert schwankt. Es ist gängige Praxis, mindestens 100-200 ns äquilibrierte Trajektorie zu erhalten, um eine angemessene statistische Analyse der interessierenden Eigenschaften und Wechselwirkungen durchzuführen. Es ist üblich, reine Membransimulationen zwischen 200 und 500 ns durchzuführen, abhängig von der Komplexität der Lipidmischung und der Forschungsfrage. Protein-Lipid-Wechselwirkungen erfordern typischerweise längere Simulationszeiten zwischen 500 und 2000 ns. Einige Ansätze zur Beschleunigung der Probenahme und der beobachtbaren Dynamik mit Membransystemen sind: (i) das hochmobile membranmimetische (HMMM) Modell, bei dem Endkohlenstoffe von Lipiden in der Membran durch organische Lösungsmittel ersetzt werden, um die Probenahme zu beschleunigen50; und (ii) Wasserstoffmassenumverteilung (HMR), die einen Bruchteil der Massen schwerer Atome innerhalb eines Systems mit denen von Wasserstoffatomen kombiniert, um die Verwendung eines größeren Simulationszeitschritts51 zu ermöglichen.
Das folgende Protokoll erläutert einen anfängerfreundlichen Ansatz zum Erstellen, Ausführen und Analysieren realistischer Membranmodelle mit AA MD. Aufgrund der Beschaffenheit von MD-Simulationen müssen mehrere Trajektorien ausgeführt werden, um die Reproduzierbarkeit und eine ordnungsgemäße statistische Analyse der Ergebnisse zu gewährleisten. Es ist gängige Praxis, mindestens drei Replikate pro relevantem System auszuführen. Sobald die Lipidspezies für den interessierenden Organismus und Prozess ausgewählt wurden, werden grundlegende Schritte zum Aufbau, Ausführen und Analysieren einer Simulationstrajektorie eines reinen Membransystems skizziert und in Abbildung 2 zusammengefasst.
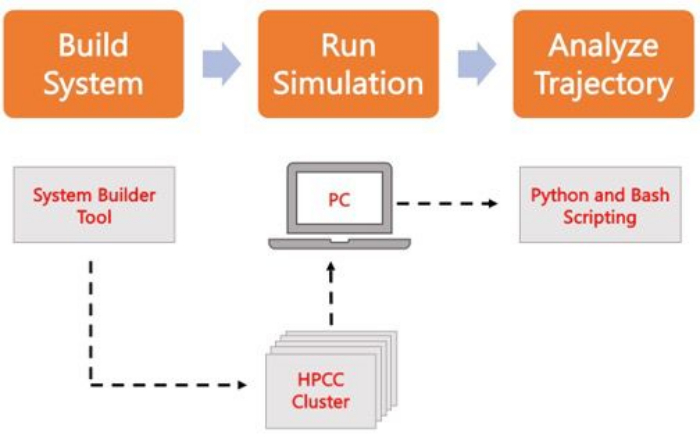
Abbildung 2. Schaltplan zum Ausführen von MD-Simulationen. Orangefarbene Kästchen entsprechen den drei Hauptschritten, die im Protokoll beschrieben sind. Darunter befindet sich der Workflow des Simulationsprozesses. Während des Systemaufbaus wird das System, das die Anfangskoordinaten eines solvatisierten Membransystems enthält, mit einem Systemeingabegenerator wie CHARMM-GUI Membrane Builder erstellt. Nach der Übertragung der Eingabedateien in einen High-Performance-Computing-Cluster wird die Simulationstrajektorie mit einer MD-Engine wie GROMACS weitergegeben. Die Trajektorienanalyse kann zusammen mit der Visualisierung auf dem Computercluster oder einer lokalen Arbeitsstation durchgeführt werden. Die Analyse wird dann entweder mithilfe von Paketen mit integriertem Analysecode wie GROMACS und VMD oder mithilfe von Bash-Skripten oder verschiedenen Python-Bibliotheken durchgeführt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
