Lipider är viktiga beståndsdelar i membran, som ger gränser för celler och möjliggör intracellulär kompartmentalisering 1,2,3. Lipider är amfifila, med en polär huvudgrupp och två hydrofoba fettsyrasvansar; Dessa självorganiserar sig i ett tvåskikt för att minimera kontakten mellan de hydrofoba kedjorna och vatten 3,4. Olika kombinationer av hydrofila huvudgrupper och hydrofoba svansar resulterar i olika klasser av lipider i biologiska membran, såsom glycerofosfolipider, sfingolipider och steroler (Figur 1)1,5,6. Glycerofosfolipider är primära byggstenar i eukaryota cellmembran som består av glycerofosfat, långkedjiga fettsyror och huvudgrupper med låg molekylvikt7. Lipidnomenklaturen baseras på skillnader i huvudgrupper; Exempel på detta är fosfatidylkolin (PC), fosfatidyletanolamin (PE), fosfatidylserin (PS), fosfatidylglycerol (PG), fosfatidylinositol (PI) eller omodifierad fosfatidinsyra (PA)5,6. När det gäller hydrofoba svansar varierar längden och mättnadsgraden, tillsammans med ryggradsstrukturen. De möjliga kombinationerna är många, vilket resulterar i tusentals lipidarter i däggdjursceller6. Förändringar i membranlipidsammansättningen leder till olika mekaniska och strukturella membranegenskaper som påverkar aktiviteten hos både integrerade membranproteiner och perifera proteiner 2,6.
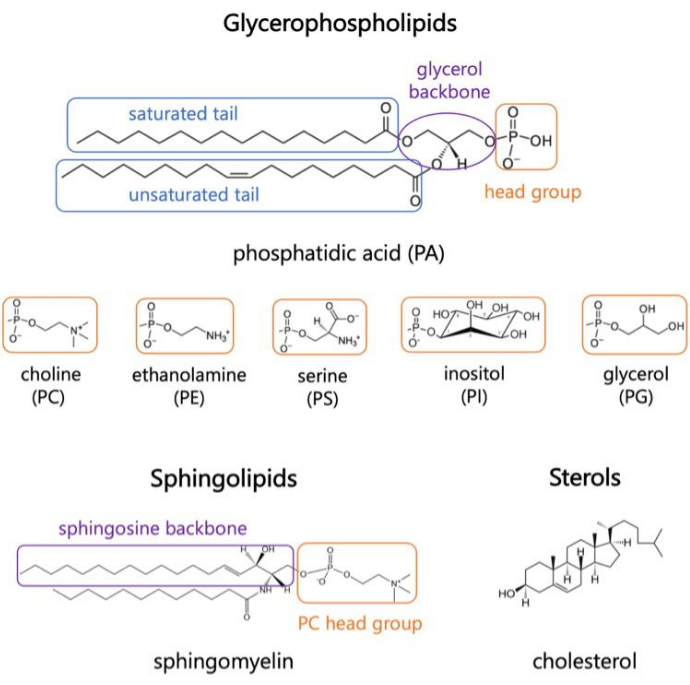
Figur 1. Representativa lipidstrukturer. Fettsyrasvansar visas i blå lådor, vanliga lipidhuvudgrupper i orange och provryggrad i lila. Klicka här för att se en större version av denna figur.
Lipider är aktiva aktörer i cellulära processer, proteinaktivering i signalkaskader och hälsosam cellhomeostas 8,9. Förändrad lipiddynamik är resultatet av infektion eller kan vara markörer för patogenes av sjukdom 10,11,12,13,14,15. Som barriärer för cellen är studiet av membranlipider och deras roll i genomträngning av små molekyler av relevans för läkemedelsleveranssystem och membranstörningsmekanismer16,17. Kemisk mångfald och olika förhållanden av lipidarter mellan organeller, vävnader och organismer ger upphov till komplex membrandynamik2. Det är därför viktigt att behålla dessa egenskaper i modelleringsstudier av lipiddubbellager, särskilt när målet med en studie är att undersöka interaktioner mellan andra biomolekyler och membranet. Vilka lipidarter som ska beaktas i en modell beror på organismen och det cellulära facket av intresse. Till exempel är PG-lipider viktiga för elektronöverföring i fotosyntetisk bateria18, medan fosforylerade inositollipider (PIP) är viktiga aktörer i plasmamembrandynamik (PM) och signalkaskader i däggdjursceller 19,20. Inuti cellen innehåller PM, endoplasmatiskt retikulum (ER), GoLgi och mitokondriella membran unika lipidöverflöd som påverkar deras funktion. Till exempel är ER navet för lipidbiogenes och transporterar kolesterol ut till PM och Golgi; den innehåller en hög lipiddiversitet med ett överflöd av PC och PE, men lågt sterolinnehåll, vilket främjar membranfluiditet21,22,23,24. Däremot innehåller PM hundratals och till och med tusentals lipidarter beroende på organismen25, den innehåller höga halter av sfingolipider och kolesterol som ger den en karakteristisk styvhet jämfört med andra membran i cellen24. Bladasymmetri bör övervägas för membran som PM, som har en yttre broschyr rik på sfingomyelin, PC och kolesterol, och en inre broschyr rik på PE, PI och PS som är viktiga för signalkaskader24. Slutligen leder lipiddiversitet också till bildandet av mikrodomäner som skiljer sig åt i packning och inre ordning, kända som lipidflottar24,26; Dessa uppvisar lateral asymmetri, antas spela viktiga roller i cellulär signalering26 och är svåra att studera på grund av deras övergående natur.
Experimentella tekniker som fluoroskopi, spektroskopi och modellmembransystem som gigantiska unilamellära vesiklar (GUV) har använts för att undersöka interaktioner mellan biomolekyler och membran. Den komplexa och dynamiska karaktären hos de inblandade komponenterna är dock svår att fånga med enbart experimentella metoder. Till exempel finns det begränsningar för avbildning av transmembrandomäner av proteiner, komplexiteten hos membran som används i sådana studier och identifiering av intermediära eller övergående tillstånd under processen av intresse27,28,29. Sedan tillkomsten av molekylär simulering av lipidmonolager och dubbellager på 1980-talet29 kan lipid-proteinsystem och deras interaktioner nu kvantifieras på molekylär nivå. Simulering av molekyldynamik (MD) är en vanlig beräkningsteknik som förutsäger partiklars rörelse baserat på deras intermolekylära krafter. En additiv interaktionspotential beskriver de bundna och icke-bundna interaktionerna mellan partiklar i systemet30. Den uppsättning parametrar som används för att modellera dessa interaktioner kallas simuleringskraftfält (FF). Dessa parametrar erhålls från ab initio-beräkningar, semi-empiriska och kvantmekaniska beräkningar och optimeras för att reproducera data från röntgen- och elektrondiffraktionsexperiment, NMR, infraröd, Raman och neutronspektroskopi, bland andra metoder31.
MD-simuleringar kan användas för att studera system på olika upplösningsnivåer32,33,34. System som syftar till att karakterisera specifika biomolekylära interaktioner, vätebindningar och andra högupplösta detaljer studeras med simuleringar av alla atomer (AA). Grovkorniga (CG) simuleringar klumpar däremot ihop atomer i större funktionella grupper för att minska beräkningskostnaderna och undersöka dynamiken i större skala33. Mellan dessa två finns UA-simuleringar (united-atoms), där väteatomer kombineras med sina respektive tunga atomer för att påskynda beräkningen33,35. MD-simuleringar är ett kraftfullt verktyg för att utforska dynamiken hos lipidmembran och deras interaktioner med andra molekyler och kan tjäna till att tillhandahålla mekanismer på molekylär nivå för processer av intresse vid membrangränssnittet. Dessutom kan MD-simuleringar användas för att begränsa experimentella mål och förutsäga makromolekylära egenskaper hos ett givet system baserat på mikroskopiska interaktioner.
I korthet, givet en uppsättning initiala koordinater, hastigheter och en uppsättning villkor som konstant temperatur och tryck, beräknas positioner och hastigheter för varje partikel genom numerisk integration av interaktionspotentialen och Newtons rörelselag. Detta upprepas iterativt och genererar därmed en simuleringsbana30. Dessa beräkningar utförs med en MD-motor; bland flera paket med öppen källkod är GROMACS36 en av de mest använda motorerna och den vi beskriver här. Den innehåller också verktyg för analys och konstruktion av initiala koordinater för system som ska simuleras37. Andra MD-motorer inkluderar NAMD38; CHARMM39 och AMBER40, som användaren kan välja efter eget gottfinnande baserat på beräkningsprestanda för ett givet system. Det är viktigt att visualisera banorna under simuleringen samt för analys och tolkning av resultaten. En mängd olika verktyg finns tillgängliga; här diskuterar vi visuell molekyldynamik (VMD) som erbjuder ett brett utbud av funktioner, inklusive tredimensionell (3-D) visualisering med expansiva ritnings- och färgläggningsmetoder, volymetrisk datavisualisering, byggande, förberedelse och analys av banor för MD-simuleringssystem och banfilmsskapande utan begränsningar för systemstorlek, om minnet är tillgängligt41,42,43.
Noggrannheten i den förutsagda dynamiken mellan systemkomponenter påverkas direkt av den FF som valts för banans utbredning. Empiriska FF-parametriseringsinsatser bedrivs av ett fåtal forskargrupper. De mest etablerade och vanliga FF för MD inkluderar CHARMM39, AMBER 40, Martini44, OPLS 45 och SIRAH 46. Kraftfältet47 för additiv CHARMM36 (C36) med alla atomer används i stor utsträckning för AA MD av membransystem eftersom det exakt reproducerar experimentella strukturdata. Den utvecklades ursprungligen av CHARMM-communityt, och den är kompatibel med flera MD-motorer som GROMACS och NAMD. Trots förbättringar i vanliga FF finns det ett kontinuerligt arbete med att förbättra parameteruppsättningarna för att möjliggöra förutsägelser som nära reproducerar experimentella observerbara värden, drivet av intressen i vissa studiesystem48,49.
En utmaning vid simulering av lipidmembran är att bestämma längden på simuleringsbanan. Detta beror till stor del på de mätvärden som ska analyseras och den process som man vill karakterisera. Vanligtvis kräver komplexa lipidblandningar längre tid för att nå jämvikt, eftersom fler arter måste ha tillräckligt med tid för att diffundera på membranplanet och nå en stabil lateral organisation. En simulering sägs vara i jämvikt när egenskapen av intresse har nått en platå och fluktuerar kring ett konstant värde. Det är vanligt att erhålla minst 100-200 ns jämviktsbana för att utföra lämplig statistisk analys av egenskaper och interaktioner av intresse. Det är vanligt att köra membransimuleringar mellan 200-500 ns, beroende på lipidblandningens komplexitet och frågeställning. Protein-lipidinteraktioner kräver vanligtvis längre simuleringstider, mellan 500-2000 ns. Några metoder för att påskynda provtagning och observerbar dynamik med membransystem är: (i) den mycket rörliga membranmimetiska (HMMM) modellen, som ersätter ändkol av lipider i membranet med organiskt lösningsmedel för att påskynda provtagning50; och (ii) vätemassepartitionering (HMR), som kombinerar en bråkdel av massan av tunga atomer i ett system med massan av väteatomer för att möjliggöra användning av ett större simuleringstidssteg51.
Följande protokoll beskriver en nybörjarvänlig metod för att bygga, köra och analysera realistiska membranmodeller med hjälp av AA MD. Med tanke på MD-simuleringarnas karaktär måste flera trajektorier köras för att ta hänsyn till reproducerbarhet och korrekt statistisk analys av resultaten. Det är aktuell praxis att köra minst tre repliker per system av intresse. När lipidarterna har valts ut för organismen och processen av intresse, beskrivs och sammanfattas grundläggande steg för att bygga, köra och analysera en simuleringsbana för ett membransystem i figur 2.
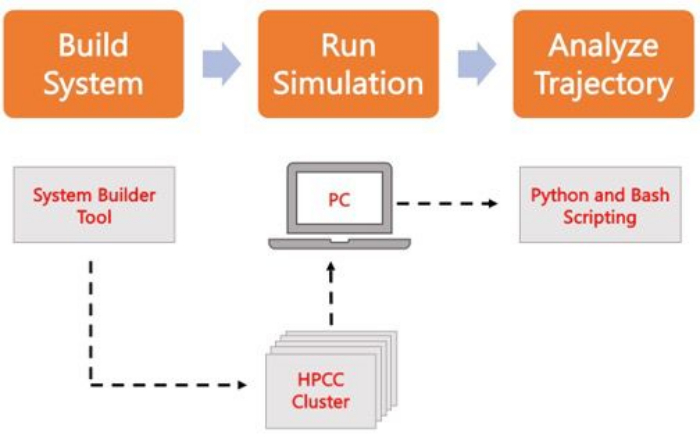
Figur 2. Schematisk för att köra MD-simuleringar. Orange rutor motsvarar de tre huvudstegen som beskrivs i protokollet. Nedanför finns arbetsflödet för simuleringsprocessen. Under systeminstallationen byggs systemet som innehåller de initiala koordinaterna för ett löst membransystem med en systemingångsgenerator som CHARMM-GUI Membrane Builder. Efter att ha överfört indatafilerna till ett högpresterande datorkluster sprids simuleringsbanan med hjälp av en MD-motor, till exempel GROMACS. Bananalys kan göras på datorklustret eller en lokal arbetsstation tillsammans med visualisering. Analysen utförs sedan antingen med hjälp av paket med inbyggd analyskod som GROMACS och VMD, eller med hjälp av Bash-skript eller olika Python-bibliotek. Klicka här för att se en större version av denna figur.
