Protein-protein-interaktioner understreger de fleste cellulære funktioner, fra immunregulering til DNA-replikation og translation. Som følge heraf er der et grundlæggende behov i hele biovidenskaben for at undersøge en lang række interaktioner på tværs af forskellige heterogene komplekser, der almindeligvis dannes. Imidlertid er deres påvisning, karakterisering og kvantificering ofte udfordrende, især for interaktioner med lav affinitet1.
Immunofældningsanalyser bruges ofte til at detektere interaktioner med høj affinitet, men for interaktioner med lav affinitet og forbigående interaktioner er detektion stort set umulig2. Fluorescensteknikker kan også anvendes, men kræver potentielt forstyrrende tilføjelse af fluorescerende etiketter2. Cryo-EM kan give et strukturelt øjebliksbillede og en ensembleaflæsning af proteinkomplekserne dannet med høj rumlig opløsning, men kræver også typisk arbejde ved koncentrationer, der er for lave til billeddannelse af lavaffinitetsinteraktioner. Cryo-EM medfører også udfordringer relateret til omkostninger, tilgængelighed, prøveforberedelse og analysetid3.
Derudover er overfladeplasmonresonans (SPR) blevet en populær måde at kvantificere protein-proteininteraktioner på, selvom det kræver proteinimmobilisering, hvilket kan påvirke bindingsligevægten og resultere i variable on-rates, hvilket reducerer målenøjagtigheden 4,5. Det involverer også flere analysetrin forud for dataindsamling og analyse6.
Massefotometri er en enkeltmolekyleteknik, der er blevet brugt til at analysere protein-proteininteraktioner 5,6,7. Det virker ved at måle massen af enkelte molekyler eller komplekser baseret på det lys, de spreder, når de lander på overfladen af et glasdæksel8. Massefotometrimålinger er blevet brugt til at kvantificere bindingsaffiniteter fra den relative overflod af bindingspartnere og de komplekser, de danner5. Ikke desto mindre bør koncentrationen af prøven, der skal måles, ligesom andre enkeltmolekyleteknikker typisk være mindre end 100 nM. Hvis koncentrationen er højere, vil molekylerne, der lander på glasoverfladen, overlappe rumligt, hvilket resulterer i dårlig datakvalitet7. Derfor kan svagere interaktioner (KD ~ mikromolarer), som dissocierer ved disse lavere koncentrationer, ikke måles pålideligt, da det ikke er muligt at observere den nødvendige blanding af ubundne og bundne arter5.
Her beskriver vi en tilgang, der overvinder denne begrænsning baseret på en ny koblet mikrofluidik massefotometrienhed. Specifikt anvendes et mikrofluidiksystem i kombination med massefotometeret til effektivt at udvide rækkevidden af interaktioner, der kan kvantificeres ved massefotometri. Mikrofluidik har vist sig at tilbyde en række muligheder for at undersøge protein-protein-interaktioner, herunder hurtig fortynding for at detektere svage interaktioner 1,9. Systemet beskrevet heri fungerer ved hurtigt at fortynde prøven op til 10.000 gange på en mikrofluidisk chip og straks strømme den over chippens observationsområde, hvilket gør det muligt for massefotometrimålingen at starte inden for 50 ms fra det tidspunkt, hvor molekylerne begyndte fortyndingsprocessen10. Fortyndingen sker, når prøven og bufferen kombineres i en omvendt Tesla-ventilblander på chippen, hvor de relative strømningshastigheder for de to opløsninger bestemmer mængden af fortynding, der forekommer (se protokoltrin 8). Strømningshastigheden kan styres med den mikrofluidiske styringssoftware. Ændring af strømningshastigheden kan ændre artens relative population, da det kan påvirke antallet af landingshændelser på overfladen af glasset, hvilket er det, der måles med massefotometeret.
Processens hastighed er hurtig nok til, at målingen kan afsluttes, før interaktionens integritet er blevet forstyrret (for yderligere detaljer, se også diskussionen). Dette kan forstås gennem et kort kig på teorien om førsteordensreaktioner, hvor 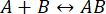 . Den fremadrettede (association) hastighedskonstant er kf, den bagudrettede (dissociation) hastighedskonstant er kb, og ligevægtsdissociationskonstanten (KD) er defineret som
. Den fremadrettede (association) hastighedskonstant er kf, den bagudrettede (dissociation) hastighedskonstant er kb, og ligevægtsdissociationskonstanten (KD) er defineret som
KD= kb/ kf
For proteinbinding er kfgenerelt begrænset af reaktanternes diffusion11 og er derfor begrænset til området 106-10 7 M-1·s-1. Fordi rækkevidden af er begrænset, vil en lavaffinitetsreaktion (KD ~ mikromolarer) have kb≈ 1 s-1. Det vil sige kb = kf · KD = (106 M-1·s-1) (10-6 M) = 1 s-1, med kompleksets halveringstid omkring 0,7 s11,12.
Vores eksempelsystem er bindingen af IgG monoklonalt antistof trastuzumab til det opløselige domæne af IgG neonatal Fc-receptoren (FcRn), som er kendte interagerende partnere13. Tidligere offentliggjorte data opnået ved hjælp af konventionel massefotometri alene (dvs. med manuel fortynding af prøver) viste, at proteinerne danner flere arter. FcRn-monomerer, FcRn-dimerer og ubundet IgG var tydeligt synlige, mens IgG-FcRn-komplekser (i forholdet 1:1 og 1:2) også blev påvist (ved pH 5,0), men kun med meget lav forekomst5. Denne observation rejser spørgsmålet om, hvorvidt IgG-FcRn-kompleksdannelse kunne detekteres tydeligere, hvis den måles ved en højere koncentration. Faktisk gav kombinationen af massefotometri med en koblet hurtig fortyndingsmetode, der er beskrevet her, mere robust bevis for kompleks dannelse ved en stigning i deres målte partikler.
Massefotometri- og mikrofluidikprotokollen beskrevet her gør det muligt at karakterisere dannelsen af komplekser med en KD op til mikromolært område. En empirisk bestemmelse af KD vil kræve yderligere forbedringer af flowsensorens nøjagtighed, pumpestabilitet, chip-til-chip-variationer og måleplacering inde i observationsvinduet, da alle disse faktorer vil påvirke tiden fra prøven fortyndes til måles.
Den samme fremgangsmåde kan anvendes til at undersøge binding mellem opløselige proteiner, forudsat at de har forskellige molekylvægte (adskilt af mindst 25 kDa), der ligger inden for det område, der er egnet til analyse med et massefotometer (30 kDa til 6 MDa). De opnåede indsigter kan være nyttige til undersøgelser i en række sammenhænge – fra at få en mekanistisk forståelse af cellulære funktioner til design af nye bioterapeutiske lægemidler.
Den her skitserede protokol giver en metode til påvisning og kvantificering af protein-protein-interaktioner med lav affinitet. Det bruger et massefotometer koblet til et mikrofluidiksystem med hurtig fortynding. Massefotometri er et etiketfrit, bioanalytisk værktøj, der pålideligt kan måle molekylmasse i opløsning til biomolekyler16 for dem inden for området 30 kDa til 6 MDa. Da massefotometri er en enkeltmolekyleteknik, der analyserer prøver en efter en, er den generelt begrænset til prøver i koncentrationsområdet 100 pM-100 nM. Over dette område vil molekylerne, der lander på glasoverfladen, overlappe rumligt, hvilket resulterer i dårlig datakvalitet; Under dette interval opnås for få data til at foretage robust analyse7. En vigtig konsekvens er, at det kan begrænse undersøgelsen af proteininteraktioner til dem, der danner en blanding af bundne og ubundne arter inden for dette interval.
Her detaljerede vi en trinvis protokol til brug af et mikrofluidiksystem med hurtig fortynding til effektivt at udvide området for prøvekoncentrationer, der er modtagelige for massefotometri. Ved at fortynde prøven på den mikrofluidiske chip og derefter strømme den over detektorobservationsvinduet inden for 50 ms fanger systemet de komplekser, der er til stede i den ufortyndede prøve, før interaktionsligevægten skifter. Prøven leveres løbende til detektoren under individuelle målinger. Under disse betingelser forbliver 95% af komplekset intakt, når prøven måles, selv for interaktioner med lav affinitet – med en KD i størrelsesordenen mikromolarer og dissociationshastigheder så hurtigt som 1 s-1.
Dette kan beregnes som følger: For en reaktion med en fremadrettet sats kfog bagudrettet hastighed kb,
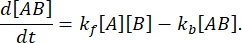
Ved ligevægt forbliver koncentrationerne af alle tre arter (A, B og komplekset AB) konstante, så 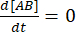 og
og 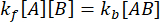 . Under den konservative antagelse, at forstyrrelsen (fortynding, i dette tilfælde) kan få komplekset til at dissociere, men den fremadrettede (association) reaktion ikke fortsætter, kan udtrykket kf [A] [B] behandles som ubetydelig, og følgende forenkling kan foretages:
. Under den konservative antagelse, at forstyrrelsen (fortynding, i dette tilfælde) kan få komplekset til at dissociere, men den fremadrettede (association) reaktion ikke fortsætter, kan udtrykket kf [A] [B] behandles som ubetydelig, og følgende forenkling kan foretages:
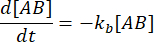
Integration giver følgende udtryk for koncentrationen af kompleks på tidspunktet efter forstyrrelsen af ligevægt:
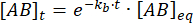
Den brøkdel af komplekset, der forbliver bundet på tidspunktet t efter forstyrrelsen af ligevægt, er således:
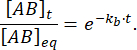
Ved = 50 ms, for en reaktion med kb≈ 1 s-1, er brøkdelen bundet 0,95 eller 95%11,12.
Massefotometri blev brugt her og tidligere5 til at undersøge bindingen af IgG monoklonalt antistof trastuzumab til det opløselige domæne af FcRn. De to bindingspartnere er rapporteret at binde med nanomolær affinitet ved sur pH17. Massefotometri blev anvendt til kvalitativt at vurdere forekomsten af de dannede komplekser, mens bindingspartnerne var ved pH 5,0, og prøverne blev hurtigt fortyndet gennem et yderligere mikrofluidisk system. Proceduren blev optimeret til den særlige protein-protein-interaktion baseret på tidligere rapporterede resultater5. Den samme procedure kan bruges til at studere andre interaktioner, forudsat at brugerne har forudgående viden eller optimerer de eksperimentelle betingelser for det pågældende system, såsom hvilke buffere der skal bruges, den indledende proteinkoncentration, den forventede støkiometri og mængden af inkubation, der er nødvendig for at tillade interaktionen at nå en ligevægt.
Når IgG-FcRn-blandingen blev fortyndet manuelt, var det vanskeligt at detektere tilstedeværelsen af IgG-FcRn-komplekser, selvom disse proteiner vides at interagere5. Dette papir viser, at den hurtige fortyndingsmetode resulterer i en markant øget mængde af disse komplekser. For den samme prøve, når hurtig fortynding blev anvendt, blev 1: 1 FcRn-IgG-komplekser og 2: 1 FcRn-IgG-komplekser begge tydeligt observeret. Disse forskelle i kompleks dannelse demonstrerer vigtigheden af at studere biomolekylære interaktionssystemer på tværs af en bred vifte af koncentrationer.
Derudover viser disse resultater også, at det er ligetil at bruge mikrofluidik med enkeltmolekyleanalyse til at fange svage interaktioner – udfylde et betydeligt hul i metoden. Kombinationen af hurtigfortyndingsmikrofluidik med massefotometri giver attraktive fordele på grund af fordelene ved massefotometri som analytisk teknik. Det vil sige, massefotometri kræver ikke etiketter, involverer minimal prøveforberedelse, og målingerne udføres i opløsning. For denne protokol er en anden vigtig fordel ved massefotometri dens evne til at skelne og kvantificere alle de dannede arter (forudsat at de har en særskilt masse på >30 kDa). Dette er i modsætning til SPR, for eksempel, som kan måle bindings- og ikke-bindingshastigheder, men ikke let kan give støkiometrioplysninger8.
For denne protokol såvel som massefotometrieksperimenter mere generelt er flere overvejelser nyttige. For det første skal den endelige proteinkoncentration være inden for grænsen for, hvad massefotometri kan måle (100 pM-100 nM). Startinkubationskoncentrationen bør også ligge inden for det mikrofluidiske systems område (op til 90 μM) og teoretiseres til at være over den faktiske KD af interaktionen10. Det anbefalede udgangspunkt er et koncentrationsblandingsforhold på 1:1 mellem de interagerende arter ved μM-koncentration. Forholdet kan derefter varieres til 1:2, 1:5 eller, som i tilfældet med denne interaktion, 1:10. Hvis der ikke er nogen tidligere oplysninger om proteininteraktionerne, skal brugeren optimere eksperimentet, begyndende med en høj koncentration (anbefalet 20 μM) for hver partner for at afgøre, om komponenternes affinitet er inden for det koncentrationsområde, der opretholdes ved den præsenterede metode (dvs. komplekser dannes). Optimering kan også omfatte valg af andre bufferbetingelser for at fremme interaktionerne eller titreringen af en af interaktionskomponenterne for at bestemme det rigtige blandingsforhold. Når disse er bestemt, er det muligt at optimere koncentrationer og strømninger, så der er optimale betingelser for undersøgelsen og metoden, f.eks. ved at sænke koncentrationerne for at muliggøre bedre topopløsning.
For det andet bør urenheder minimeres for at kunne replikere dette eksperiment. Almindelige kilder til urenheder, der vides at påvirke massefotometrimålinger negativt, omfatter andre proteiner eller cellulært affald, der forbliver efter rensning, ufiltrerede buffere, micelledannende rengøringsmidler (hvis de er til stede i for høj koncentration) og buffere indeholdende høje koncentrationer af salt, glycerol eller andre komponenter. Som diskuteret i protokollen ovenfor bør bobler i mikrofluidiksystemet fjernes. Der kan dannes bobler i slangesystemet, eller hvis prøverne har høj overfladespænding og er tilbøjelige til skumdannelse. Der kan også dannes bobler i nedsænkningsolien, som kan detekteres fra fokusringen (figur 3). Hvis bobler ikke kan fjernes ved hjælp af de trin, der er beskrevet i protokollen, er en anden løsning at afgasse prøven ved hjælp af en ekssikkator og en vakuumpumpe, så prøven efterlades under reduceret tryk i et par minutter. Vortexing eller rystning af stærkt koncentrerede proteinopløsninger anbefales ikke, da disse handlinger kan fremme bobledannelse.
Mens målingen af en specifik protein-protein-interaktion demonstreres her, kan den samme protokol anvendes på andre protein-protein-interaktionssystemer uden væsentlig ændring. En yderligere fremtidig retning for denne protokol ville være at bruge målingerne til at beregne KD-værdier for de identificerede komplekser, som det er beskrevet andetsteds i forbindelse med massefotometri 5,7. Mens de tidligere undersøgelser anvendte data fra eksperimenter, der involverede manuel fortynding og stærkere interaktioner, kunne analyseprincippet let anvendes i denne sammenhæng – forudsat at der implementeres yderligere forbedringer i den mikrofluidiske enhed (såsom øget flowsensornøjagtighed og pumpestabilitet).
Ud over protein-protein-interaktioner vil der sandsynligvis være bredere anvendelser for den kombinerede massefotometri og hurtigfortyndingsmikrofluidik-tilgang. Massefotometri kan bruges til at vurdere prøvens renhed, aggregering og homogenitet18,19; undersøgelse af proteinoligomerisering20, makromolekylær samling21 eller polymerisering22; og på andre områder. Massefotometrianalyse strækker sig også ud over proteiner; Det er blevet brugt til at undersøge interaktioner mellem nukleinsyrer og proteiner23, virale partikler24 og nanopartikler25. Denne protokol beskriver således en vigtig anvendelse af et kombineret massefotometrimikrofluidiksystem – det muliggør direkte måling af svage protein-proteininteraktioner på niveau med individuelle molekyler og komplekser. Værdien af denne anvendelse er høj, da den åbner mulighed for ligefrem at karakterisere interaktioner, der generelt har været vanskelige at studere – med relevans på tværs af kritiske terapeutiske områder. Denne kombinerede tilgang kunne også tjene som grundlag for en bredere vifte af undersøgelser af prøver med koncentrationer op til snesevis af mikromolarer.
