Las interacciones proteína-proteína subrayan la mayoría de las funciones celulares, desde la regulación inmunitaria hasta la replicación y traducción del ADN. Como resultado, existe una necesidad fundamental en todas las ciencias de la vida para investigar una amplia gama de interacciones a través de diversos complejos heterogéneos que se forman comúnmente. Sin embargo, su detección, caracterización y cuantificación suelen ser un reto, especialmente en el caso de las interacciones de baja afinidad1.
Los ensayos de inmunoprecipitación se utilizan a menudo para detectar interacciones de alta afinidad, pero para interacciones transitorias y de baja afinidad, la detección es en gran medida inviable2. También se pueden utilizar técnicas de fluorescencia, pero requieren la adición potencialmente disruptiva de etiquetasfluorescentes 2. Cryo-EM puede proporcionar una instantánea estructural y una lectura de conjunto de los complejos de proteínas formados con alta resolución espacial, pero también suele requerir trabajar a concentraciones que son demasiado bajas para obtener imágenes de interacciones de baja afinidad. Cryo-EM también presenta desafíos relacionados con el costo, la accesibilidad, la preparación de muestras y el tiempo de análisis3.
Además, la resonancia de plasmón de superficie (SPR) se ha convertido en una forma popular de cuantificar las interacciones proteína-proteína, aunque requiere la inmovilización de proteínas, lo que puede afectar el equilibrio de unión y dar lugar a tasas de activación variables, reduciendo así la precisión de la medición 4,5. También implica varios pasos de ensayo antes de la recopilación y el análisis de datos6.
La fotometría masiva es una técnica de una sola molécula que se ha utilizado para analizar las interacciones proteína-proteína 5,6,7. Funciona midiendo la masa de moléculas individuales o complejos en función de la luz que dispersan cuando aterrizan en la superficie de un cubreobjetos de vidrio8. Las mediciones de fotometría masiva se han utilizado para cuantificar las afinidades de unión a partir de la abundancia relativa de los socios de unión y los complejos que forman5. Sin embargo, al igual que otras técnicas de una sola molécula, la concentración de la muestra que se va a medir suele ser inferior a 100 nM. Si la concentración es mayor, las moléculas que aterrizan en la superficie del vidrio se superpondrán espacialmente, lo que dará como resultado una mala calidad de los datos7. En consecuencia, las interacciones más débiles (KD ~ micromolares), que se disocian a estas concentraciones más bajas, no pueden medirse de manera confiable ya que no es posible observar la mezcla necesaria de especies unidas y no unidas5.
Aquí, describimos un enfoque que supera esta limitación basado en un nuevo dispositivo de fotometría masiva de microfluídica acoplada. Específicamente, un sistema de microfluídica se utiliza en combinación con el fotómetro de masas para expandir de manera efectiva la gama de interacciones que se pueden cuantificar mediante fotometría de masas. Se ha demostrado que la microfluídica ofrece una gama de posibilidades para investigar las interacciones proteína-proteína, incluida la dilución rápida para detectar interacciones débiles 1,9. El sistema descrito en este documento funciona diluyendo rápidamente la muestra hasta 10.000 veces en un chip microfluídico y fluyendo inmediatamente a través del área de observación del chip, lo que permite que la medición de la fotometría de masas comience dentro de los 50 ms desde que las moléculas comenzaron el proceso de dilución10. La dilución se produce cuando la muestra y el tampón se combinan en un mezclador de válvula Tesla inversa en el chip, y los caudales relativos de las dos soluciones determinan la cantidad de dilución que se produce (consulte el paso 8 del protocolo). El caudal es controlable con el software de control microfluídico. La alteración del caudal puede cambiar la población relativa de la especie, ya que puede afectar el número de eventos de aterrizaje en la superficie del vidrio, que es lo que mide el fotómetro de masa.
La velocidad del proceso es lo suficientemente rápida como para que la medición se complete antes de que se interrumpa la integridad de la interacción (para más detalles, consulte también la Discusión). Esto se puede entender a través de una breve mirada a la teoría de las reacciones de primer orden, donde 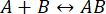 . La constante de velocidad hacia adelante (asociación) es kf, la constante de tasa hacia atrás (disociación) es kb y la constante de disociación de equilibrio (KD) se define como
. La constante de velocidad hacia adelante (asociación) es kf, la constante de tasa hacia atrás (disociación) es kb y la constante de disociación de equilibrio (KD) se define como
KD= kb/ kf
Para la unión a proteínas, kfestá generalmente limitada por la difusión de los reactivos11 y, por lo tanto, está restringida al rango de 10 6-107 M-1·s-1. Debido a que el rango de es limitado, una reacción de baja afinidad (KD ~ micromolares) tendrá kb≈ 1 s-1. Es decir, kb= kf · KD= (106 M-1·s-1) (10-6 M) = 1 s-1, con una vida media del complejo de alrededor de 0,7 s11,12.
Nuestro sistema de ejemplo es la unión del anticuerpo monoclonal IgG trastuzumab al dominio soluble del receptor Fc neonatal de IgG (FcRn), que se sabe que interactúan13. Los datos publicados anteriormente obtenidos utilizando solo fotometría masiva convencional (es decir, con dilución manual de muestras) mostraron que las proteínas forman múltiples especies. Los monómeros de FcRn, los dímeros de FcRn y la IgG no unida fueron claramente visibles, mientras que los complejos IgG-FcRn (en proporciones 1:1 y 1:2) también se detectaron (a pH 5,0) pero solo con una abundancia muy baja5. Esta observación plantea la cuestión de si la formación del complejo IgG-FcRn podría detectarse más claramente si se mide a una concentración más alta. De hecho, la combinación de la fotometría masiva con un enfoque de dilución rápida acoplada descrito aquí proporcionó pruebas más sólidas de la formación de complejos mediante un aumento de las partículas medidas.
El protocolo de fotometría de masas y microfluídica aquí descrito permite caracterizar la formación de complejos con un KD hasta el rango micromolar. Una determinación empírica del KD requerirá mejoras adicionales en la precisión del sensor de flujo, la estabilidad de la bomba, las variaciones de viruta a viruta y la ubicación de la medición dentro de la ventana de observación, ya que todos estos factores influirían en el tiempo desde que la muestra se diluye hasta que se mide.
El mismo enfoque podría aplicarse para investigar la unión entre cualquier proteína soluble, siempre que tenga pesos moleculares distintos (separados por al menos 25 kDa) que se encuentren en el rango adecuado para el análisis con un fotómetro de masa (30 kDa a 6 MDa). Los conocimientos obtenidos podrían ser útiles para estudios en una variedad de contextos, desde la obtención de una comprensión mecanicista de las funciones celulares hasta el diseño de nuevos medicamentos bioterapéuticos.
El protocolo descrito aquí proporciona un método para detectar y cuantificar las interacciones proteína-proteína de baja afinidad. Utiliza un fotómetro de masas acoplado a un sistema de microfluídica de dilución rápida. La fotometría de masas es una herramienta bioanalítica sin marcadores que puede medir de forma fiable la masa molecular en solución para biomoléculas16, para aquellas dentro del rango de 30 kDa a 6 MDa. Como la fotometría de masas es una técnica de una sola molécula que analiza las muestras una por una, generalmente se limita a muestras en el rango de concentración de 100 pM-100 nM. Por encima de este rango, las moléculas que aterrizan en la superficie del vidrio se superpondrán espacialmente, lo que dará lugar a una mala calidad de los datos; Por debajo de este rango, se obtienen muy pocos datos para realizar un análisis robusto7. Una consecuencia importante es que puede limitar la investigación de las interacciones de las proteínas a aquellas que forman una mezcla de especies unidas y no unidas dentro de ese rango.
Aquí, detallamos un protocolo paso a paso para usar un sistema de microfluídica de dilución rápida para expandir de manera efectiva el rango de concentraciones de muestras que son susceptibles de fotometría masiva. Al diluir la muestra en el chip microfluídico y luego hacerla fluir a través de la ventana de observación del detector dentro de los 50 ms, el sistema captura los complejos presentes en la muestra sin diluir antes de que cambie el equilibrio de interacción. La muestra se suministra continuamente al detector durante las mediciones individuales. En estas condiciones, el 95% del complejo permanecerá intacto cuando se mida la muestra, incluso para interacciones de baja afinidad, con un KD del orden de los micromolares y tasas de disociación tan rápidas como 1 s-1.
Esto se puede calcular de la siguiente manera: Para una reacción con una velocidad hacia adelante kfy una velocidad hacia atrás kb,
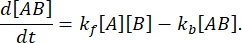
En equilibrio, las concentraciones de las tres especies (A, B y el complejo AB) permanecen constantes, por lo que 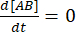 y
y 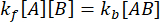 . Bajo la suposición conservadora de que la perturbación (dilución, en este caso) puede hacer que el complejo se disocie, pero la reacción directa (asociación) no procede, el término kf [A] [B] puede tratarse como despreciable, y se puede hacer la siguiente simplificación:
. Bajo la suposición conservadora de que la perturbación (dilución, en este caso) puede hacer que el complejo se disocie, pero la reacción directa (asociación) no procede, el término kf [A] [B] puede tratarse como despreciable, y se puede hacer la siguiente simplificación:
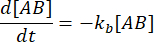
La integración da la siguiente expresión para la concentración de complejo en el tiempo después de la perturbación del equilibrio:
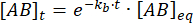
La fracción del complejo que permanece ligada en el tiempo t después de la perturbación del equilibrio es así:
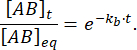
A = 50 ms, para una reacción con kb≈ 1 s-1, el enlace de la fracción es 0,95, o 95%11,12.
La fotometría masiva se utilizó aquí y anteriormente5 para investigar la unión del anticuerpo monoclonal IgG trastuzumab al dominio soluble del FcRn. Se ha informado que los dos socios de unión se unen con afinidad nanomolar a pHácido 17. Se utilizó fotometría masiva para evaluar cualitativamente la abundancia de los complejos formados mientras los socios de unión estaban a pH 5.0, y las muestras se diluyeron rápidamente a través de un sistema microfluídico adicional. El procedimiento fue optimizado para la interacción proteína-proteína particular con base en los resultados reportados previamente5. El mismo procedimiento se puede utilizar para estudiar otras interacciones, siempre que los usuarios tengan conocimientos previos u optimicen las condiciones experimentales para el sistema en cuestión, como qué tampones utilizar, la concentración inicial de proteínas, la estequiometría esperada y la cantidad de incubación necesaria para permitir que la interacción alcance un equilibrio.
Cuando la mezcla IgG-FcRn se diluyó manualmente, fue difícil detectar la presencia de complejos IgG-FcRn, a pesar de que se sabe que estas proteínas interactúan5. Este trabajo muestra que el enfoque de dilución rápida da como resultado una cantidad notablemente mayor de estos complejos. Para la misma muestra, cuando se utilizó una dilución rápida, se observaron claramente complejos FcRn-IgG 1:1 y 2:1 FcRn-IgG. Estas diferencias en la formación de complejos demuestran la importancia de estudiar los sistemas de interacción biomolecular en una amplia gama de concentraciones.
Además, estos resultados también demuestran que es sencillo utilizar la microfluídica con el análisis de una sola molécula para capturar interacciones débiles, llenando un vacío significativo en el método. La combinación de la microfluídica de dilución rápida con la fotometría de masas ofrece ventajas atractivas debido a las ventajas de la fotometría de masas como técnica analítica. Es decir, la fotometría masiva no requiere etiquetas, implica una preparación mínima de la muestra y las mediciones se realizan en solución. Para este protocolo, otra ventaja clave de la fotometría masiva es su capacidad para distinguir y cuantificar todas las especies formadas (siempre que tengan una masa distinta de >30 kDa). Esto contrasta con la SPR, por ejemplo, que puede medir las tasas de unión y desunión, pero no puede proporcionar fácilmente información estequiométrica8.
Para este protocolo, así como para los experimentos de fotometría masiva en general, son útiles varias consideraciones. En primer lugar, la concentración final de proteínas debe estar dentro del límite de lo que la fotometría de masas puede medir (100 pM-100 nM). La concentración de incubación inicial también debe estar dentro del rango del sistema microfluídico (hasta 90 μM) y se teoriza que está por encima del KD real de la interacción10. El punto de partida recomendado es una proporción de mezcla de concentración de 1:1 entre las especies que interactúan a una concentración de μM. La relación podría entonces variarse a 1:2, 1:5 o, como en el caso de esta interacción, 1:10. Si no hay información previa sobre las interacciones de las proteínas, el usuario tendría que optimizar el experimento, comenzando con una concentración alta (recomendada 20 μM) para cada socio para determinar si la afinidad de los componentes está dentro del rango de concentración sostenido por el método presentado (es decir, se forman complejos). La optimización también puede implicar la elección de otras condiciones de búfer para promover las interacciones o la valoración de uno de los componentes de interacción para determinar la proporción de mezcla correcta. Una vez determinados, es posible optimizar las concentraciones y los caudales para permitir condiciones óptimas para el estudio y el método, por ejemplo, disminuyendo las concentraciones para permitir una mejor resolución de los picos.
En segundo lugar, para replicar con éxito este experimento, se deben minimizar las impurezas. Las fuentes comunes de impurezas que se sabe que afectan negativamente a las mediciones de fotometría masiva incluyen otras proteínas o desechos celulares que permanecen después de la purificación, tampones sin filtrar, detergentes formadores de micelas (si están presentes en una concentración demasiado alta) y tampones que contienen altas concentraciones de sal, glicerol u otros componentes. Como se discutió en el Protocolo anterior, las burbujas en el sistema microfluídico deben eliminarse. Se pueden formar burbujas en el sistema de tubos o si las muestras tienen una alta tensión superficial y son propensas a la formación de espuma. También se pueden formar burbujas en el aceite de inmersión, que se pueden detectar desde el anillo de enfoque (Figura 3). Si las burbujas no se pueden eliminar siguiendo los pasos descritos en el protocolo, otra solución es desgasificar la muestra con un desecador y una bomba de vacío, dejando la muestra a presión reducida durante unos minutos. No se recomienda agitar o agitar soluciones de proteínas altamente concentradas, ya que estas acciones pueden promover la formación de burbujas.
Si bien aquí se demuestra la medición de una interacción proteína-proteína específica, el mismo protocolo se puede aplicar a otros sistemas de interacción proteína-proteína sin modificaciones significativas. Otra dirección futura de este protocolo sería utilizar las mediciones para calcular los valores deK D para los complejos identificados, como se ha descrito en otro lugar en el contexto de la fotometría de masas 5,7. Si bien los estudios anteriores utilizaron datos de experimentos que involucraban dilución manual e interacciones más fuertes, el principio de análisis podría aplicarse fácilmente en este contexto, siempre que se implementen mejoras adicionales en el dispositivo microfluídico (como una mayor precisión del sensor de flujo y estabilidad de la bomba).
Más allá de las interacciones proteína-proteína, es probable que haya aplicaciones más amplias para el enfoque combinado de fotometría de masas y microfluídica de dilución rápida. La fotometría masiva se puede utilizar para evaluar la pureza, agregación y homogeneidad de la muestra 18,19; estudiar la oligomerizaciónde proteínas 20, el ensamblaje macromolecular21 o la polimerización22; y en otros ámbitos. El análisis de fotometría masiva también se extiende más allá de las proteínas; Se ha utilizado para investigar las interacciones entre los ácidos nucleicos y las proteínas23, las partículas virales24 y las nanopartículas25. Por lo tanto, este protocolo describe una aplicación importante de un sistema combinado de microfluídica de fotometría de masas: permite la medición directa de interacciones débiles proteína-proteína a nivel de moléculas y complejos individuales. El valor de la presente aplicación es alto, ya que abre la posibilidad de caracterizar directamente interacciones que generalmente han sido difíciles de estudiar, con relevancia en áreas terapéuticas críticas. Este enfoque combinado también podría servir como base para una gama más amplia de investigaciones para muestras con concentraciones de hasta decenas de micromolares.
