Protein-Protein-Wechselwirkungen unterstreichen die meisten zellulären Funktionen, von der Immunregulation über die DNA-Replikation bis hin zur Translation. Infolgedessen besteht in den Lebenswissenschaften ein grundlegender Bedarf, ein breites Spektrum an Wechselwirkungen zwischen verschiedenen heterogenen Komplexen zu untersuchen, die häufig gebildet werden. Ihre Erkennung, Charakterisierung und Quantifizierung sind jedoch oft eine Herausforderung, insbesondere bei Wechselwirkungen mit geringer Affinität1.
Immunpräzipitationsassays werden häufig zum Nachweis von Wechselwirkungen mit hoher Affinität verwendet, aber bei Wechselwirkungen mit niedriger Affinität und transienten Wechselwirkungen ist ein Nachweis weitgehend nicht möglich2. Fluoreszenztechniken können ebenfalls verwendet werden, erfordern jedoch die potenziell störende Zugabe von Fluoreszenzmarkierungen2. Kryo-EM kann eine strukturelle Momentaufnahme und eine Ensemble-Auslesung der gebildeten Proteinkomplexe mit hoher räumlicher Auflösung liefern, erfordert aber in der Regel auch die Arbeit mit Konzentrationen, die für die Abbildung von Wechselwirkungen mit geringer Affinität zu niedrig sind. Die Kryo-EM bringt auch Herausforderungen in Bezug auf Kosten, Zugänglichkeit, Probenvorbereitung und Analysezeitmit sich 3.
Darüber hinaus hat sich die Oberflächenplasmonenresonanz (SPR) zu einer beliebten Methode zur Quantifizierung von Protein-Protein-Wechselwirkungen entwickelt, obwohl sie eine Proteinimmobilisierung erfordert, die das Bindungsgleichgewicht beeinträchtigen und zu variablen On-Raten führen kann, wodurch die Messgenauigkeit verringertwird 4,5. Es umfasst auch mehrere Analyseschritte vor der Datenerfassung und -analyse6.
Die Massenphotometrie ist eine Einzelmolekültechnik, die zur Analyse von Protein-Protein-Wechselwirkungen verwendet wurde 5,6,7. Es misst die Masse einzelner Moleküle oder Komplexe auf der Grundlage des Lichts, das sie streuen, wenn sie auf der Oberfläche eines Glasdeckglases8 landen. Massenphotometrische Messungen wurden verwendet, um die Bindungsaffinitäten aus der relativen Häufigkeit der Bindungspartner und den von ihnen gebildeten Komplexen zu quantifizieren5. Wie bei anderen Einzelmolekültechniken sollte die Konzentration der zu messenden Probe jedoch in der Regel weniger als 100 nM betragen. Ist die Konzentration höher, überlappen sich die Moleküle, die auf der Glasoberfläche landen, räumlich, was zu einer schlechten Datenqualität führt7. Folglich können schwächere Wechselwirkungen (KD ~ Mikromolaren), die bei diesen niedrigeren Konzentrationen dissoziieren, nicht zuverlässig gemessen werden, da es nicht möglich ist, die notwendige Mischung aus ungebundenen und gebundenen Spezieszu beobachten 5.
Hier beschreiben wir einen Ansatz, der diese Einschränkung auf der Grundlage eines neuen gekoppelten Mikrofluidik-Massenphotometriegeräts überwindet. Konkret wird ein Mikrofluidik-System in Kombination mit dem Massenphotometer eingesetzt, um den Bereich der Wechselwirkungen, die durch Massenphotometrie quantifiziert werden können, effektiv zu erweitern. Es hat sich gezeigt, dass die Mikrofluidik eine Reihe von Möglichkeiten zur Untersuchung von Protein-Protein-Wechselwirkungen bietet, einschließlich einer schnellen Verdünnung zum Nachweis schwacher Wechselwirkungen 1,9. Das hierin beschriebene System funktioniert, indem es die Probe auf einem mikrofluidischen Chip schnell bis zu 10.000-fach verdünnt und sie sofort über den Beobachtungsbereich des Chips fließen lässt, wodurch die Massenphotometriemessung innerhalb von 50 ms nach dem Zeitpunkt beginnen kann, an dem die Moleküle den Verdünnungsprozess10 begonnen haben. Die Verdünnung tritt auf, wenn die Probe und der Puffer in einem umgekehrten Tesla-Ventilmischer auf dem Chip kombiniert werden, wobei die relativen Durchflussraten der beiden Lösungen die Menge der auftretenden Verdünnung bestimmen (siehe Protokollschritt 8). Die Durchflussmenge ist mit der mikrofluidischen Steuerungssoftware steuerbar. Eine Änderung der Durchflussrate kann die relative Population der Spezies verändern, da sie die Anzahl der Landeereignisse auf der Oberfläche des Glases beeinflussen kann, die vom Massenphotometer gemessen wird.
Die Geschwindigkeit des Prozesses ist schnell genug, um die Messung abzuschließen, bevor die Integrität der Wechselwirkung gestört wurde (für weitere Details siehe auch die Diskussion). Dies kann durch einen kurzen Blick auf die Theorie der Reaktionen erster Ordnung verstanden werden, wobei 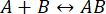 . Die Vorwärts-(Assoziations-)Geschwindigkeitskonstante ist kf, die Rückwärts-Ratenkonstante (Dissoziations-) ist kb und die Gleichgewichtsdissoziationskonstante (KD) ist definiert als
. Die Vorwärts-(Assoziations-)Geschwindigkeitskonstante ist kf, die Rückwärts-Ratenkonstante (Dissoziations-) ist kb und die Gleichgewichtsdissoziationskonstante (KD) ist definiert als
KD= kb/ kf
Für die Proteinbindung ist kfim Allgemeinen durch die Diffusion11 der Reaktanten begrenzt und daher auf den Bereich von 106-10 7 M-1·s-1 beschränkt. Da der Bereich von begrenzt ist, wird eine Reaktion mit niedriger Affinität (KD~Mikromolaren) kb≈ 1 s-1 aufweisen. Das heißt, kb= kf · KD= (106 M-1·s-1) (10-6 M) = 1 s-1, mit einer Halbwertszeit des Komplexes von etwa 0,7 s11,12.
Unser Beispielsystem ist die Bindung des monoklonalen IgG-Antikörpers Trastuzumab an die lösliche Domäne des neonatalen IgG-Fc-Rezeptors (FcRn), die als Interaktionspartner bekannt sind13. Zuvor veröffentlichte Daten, die allein mit konventioneller Massenphotometrie (d.h. mit manueller Verdünnung der Proben) gewonnen wurden, zeigten, dass die Proteine mehrere Spezies bilden. FcRn-Monomere, FcRn-Dimere und ungebundenes IgG waren deutlich sichtbar, während IgG-FcRn-Komplexe (im Verhältnis 1:1 und 1:2) ebenfalls nachgewiesen wurden (bei pH 5,0), jedoch nur mit sehr geringer Häufigkeit5. Diese Beobachtung wirft die Frage auf, ob die Bildung des IgG-FcRn-Komplexes deutlicher nachgewiesen werden kann, wenn sie in einer höheren Konzentration gemessen wird. Tatsächlich lieferte die Kombination von Massenphotometrie mit einem hier beschriebenen gekoppelten Ansatz der schnellen Verdünnung einen robusteren Beweis für die Komplexbildung durch eine Zunahme ihrer gemessenen Partikel.
Das hier beschriebene Protokoll der Massenphotometrie und Mikrofluidik ermöglicht es, die Bildung von Komplexen mit einem KD bis in den mikromolaren Bereich zu charakterisieren. Eine empirische Bestimmung der KD erfordert weitere Verbesserungen der Genauigkeit des Durchflusssensors, der Pumpenstabilität, der Span-zu-Chip-Variationen und des Messortes innerhalb des Beobachtungsfensters, da all diese Faktoren die Zeit von der Verdünnung der Probe bis zur Messung beeinflussen würden.
Der gleiche Ansatz könnte angewendet werden, um die Bindung zwischen löslichen Proteinen zu untersuchen, vorausgesetzt, sie haben unterschiedliche Molekulargewichte (mindestens 25 kDa getrennt), die in den für die Analyse mit einem Massenphotometer geeigneten Bereich (30 kDa bis 6 MDa) fallen. Die gewonnenen Erkenntnisse könnten für Studien in einer Reihe von Kontexten hilfreich sein – vom mechanistischen Verständnis zellulärer Funktionen bis hin zum Design neuer biotherapeutischer Medikamente.
Das hier skizzierte Protokoll bietet eine Methode zum Nachweis und zur Quantifizierung von Protein-Protein-Wechselwirkungen mit geringer Affinität. Es verwendet ein Massenphotometer, das mit einem Mikrofluidiksystem mit schneller Verdünnung gekoppelt ist. Die Massenphotometrie ist ein markierungsfreies, bioanalytisches Werkzeug, mit dem die Molekülmasse in Lösung für Biomoleküle16 zuverlässig gemessen werden kann, für solche im Bereich von 30 kDa bis 6 MDa. Da es sich bei der Massenphotometrie um eine Einzelmolekültechnik handelt, bei der Proben einzeln analysiert werden, ist sie im Allgemeinen auf Proben im Konzentrationsbereich von 100 pM bis 100 nM beschränkt. Oberhalb dieses Bereichs überlappen sich die Moleküle, die auf der Glasoberfläche landen, räumlich, was zu einer schlechten Datenqualität führt. Unterhalb dieses Bereichs werden zu wenige Daten für eine robuste Analyse gewonnen7. Eine wichtige Konsequenz ist, dass die Untersuchung von Proteininteraktionen auf solche beschränkt werden kann, die innerhalb dieses Bereichs eine Mischung aus gebundenen und ungebundenen Spezies bilden.
Hier haben wir ein Schritt-für-Schritt-Protokoll für die Verwendung eines Mikrofluidiksystems mit schneller Verdünnung beschrieben, um den Bereich der Probenkonzentrationen, die für die Massenphotometrie geeignet sind, effektiv zu erweitern. Indem die Probe auf dem mikrofluidischen Chip verdünnt und dann innerhalb von 50 ms über das Beobachtungsfenster des Detektors geleitet wird, erfasst das System die in der unverdünnten Probe vorhandenen Komplexe, bevor sich das Wechselwirkungsgleichgewicht verschiebt. Die Probe wird während einzelner Messungen kontinuierlich dem Detektor zugeführt. Unter diesen Bedingungen bleiben 95 % des Komplexes intakt, wenn die Probe gemessen wird, selbst bei Wechselwirkungen mit geringer Affinität – mit einem KD in der Größenordnung von Mikromolaren und Dissoziationsraten von nur 1 s-1.
Dies kann wie folgt berechnet werden: Für eine Reaktion mit einer Vorwärtsrate kfund einer Rückwärtsrate kb
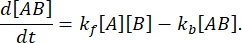
Im Gleichgewicht bleiben die Konzentrationen aller drei Spezies (A, B und der Komplex AB) konstant, also 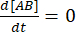 und
und 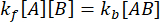 . Unter der konservativen Annahme, dass die Störung (in diesem Fall Verdünnung) dazu führen kann, dass der Komplex dissoziiert, die Vorwärtsreaktion (Assoziationsreaktion) jedoch nicht abläuft, kann der Term kf [A] [B] als vernachlässigbar behandelt werden, und es kann die folgende Vereinfachung vorgenommen werden:
. Unter der konservativen Annahme, dass die Störung (in diesem Fall Verdünnung) dazu führen kann, dass der Komplex dissoziiert, die Vorwärtsreaktion (Assoziationsreaktion) jedoch nicht abläuft, kann der Term kf [A] [B] als vernachlässigbar behandelt werden, und es kann die folgende Vereinfachung vorgenommen werden:
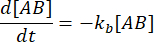
Die Integration ergibt den folgenden Ausdruck für die Konzentration des Komplexes zum Zeitpunkt nach der Störung des Gleichgewichts:
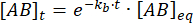
Der Anteil des Komplexes, der zum Zeitpunkt t nach der Gleichgewichtsstörung noch gebunden bleibt, ist also:
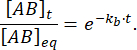
Bei = 50 ms beträgt für eine Reaktion mit kb≈ 1 s-1 die gebundene Fraktion 0,95 oder 95%11,12.
Die Massenphotometrie wurde hier und zuvor5 eingesetzt, um die Bindung des monoklonalen IgG-Antikörpers Trastuzumab an die lösliche Domäne des FcRn zu untersuchen. Es wurde berichtet, dass die beiden Bindungspartner bei saurem pH17 mit nanomolarer Affinität binden. Die Massenphotometrie wurde verwendet, um die Häufigkeit der gebildeten Komplexe qualitativ zu beurteilen, während die Bindungspartner einen pH-Wert von 5,0 hatten, und die Proben wurden durch ein zusätzliches mikrofluidisches System schnell verdünnt. Das Verfahren wurde auf der Grundlage der zuvor berichteten Ergebnisse für die jeweilige Protein-Protein-Interaktion optimiert5. Das gleiche Verfahren kann verwendet werden, um andere Wechselwirkungen zu untersuchen, vorausgesetzt, die Benutzer verfügen über Vorkenntnisse oder optimieren die experimentellen Bedingungen für das betreffende System, z. B. welche Puffer verwendet werden sollen, die anfängliche Proteinkonzentration, die erwartete Stöchiometrie und die Menge an Inkubation, die erforderlich ist, damit die Interaktion ein Gleichgewicht erreicht.
Wenn das IgG-FcRn-Gemisch manuell verdünnt wurde, war es schwierig, das Vorhandensein von IgG-FcR-Komplexen nachzuweisen, obwohl bekannt ist, dass diese Proteine interagieren5. Diese Arbeit zeigt, dass der Ansatz der schnellen Verdünnung zu einer deutlich erhöhten Menge dieser Komplexe führt. Bei derselben Probe wurden bei Verwendung einer Schnellverdünnung sowohl 1:1 FcRn-IgG-Komplexe als auch 2:1 FcRn-IgG-Komplexe deutlich beobachtet. Diese Unterschiede in der Komplexbildung zeigen, wie wichtig es ist, biomolekulare Interaktionssysteme über einen breiten Konzentrationsbereich hinweg zu untersuchen.
Darüber hinaus zeigen diese Ergebnisse auch, dass es einfach ist, Mikrofluidik mit Einzelmolekülanalyse zu verwenden, um schwache Wechselwirkungen zu erfassen – und damit eine bedeutende Lücke in der Methode zu schließen. Die Kombination der Mikrofluidik mit der Massenphotometrie bietet aufgrund der Vorteile der Massenphotometrie als Analysetechnik attraktive Vorteile. Das heißt, die Massenphotometrie erfordert keine Markierungen, erfordert nur eine minimale Probenvorbereitung und die Messungen werden in Lösung durchgeführt. Ein weiterer wichtiger Vorteil der Massenphotometrie für dieses Protokoll ist ihre Fähigkeit, alle gebildeten Spezies zu unterscheiden und zu quantifizieren (vorausgesetzt, sie haben eine ausgeprägte Masse von >30 kDa). Dies steht im Gegensatz zur SPR, die beispielsweise die Bindungs- und Entbindungsraten messen kann, aber keine Stöchiometrie-Informationen liefern kann8.
Sowohl für dieses Protokoll als auch für Massenphotometrie-Experimente im Allgemeinen sind mehrere Überlegungen hilfreich. Zunächst sollte die endgültige Proteinkonzentration innerhalb der Grenze dessen liegen, was die Massenphotometrie messen kann (100 pM-100 nM). Die Anfangsinkubationskonzentration sollte ebenfalls innerhalb des Bereichs des mikrofluidischen Systems (bis zu 90 μM) liegen und theoretisch über dem tatsächlichen KD der Wechselwirkung10 liegen. Der empfohlene Ausgangspunkt ist ein Konzentrationsmischungsverhältnis von 1:1 zwischen den wechselwirkenden Spezies bei μM-Konzentration. Das Verhältnis könnte dann auf 1:2, 1:5 oder, wie im Fall dieser Wechselwirkung, 1:10 variiert werden. Wenn es keine vorherigen Informationen über die Proteininteraktionen gibt, müsste der Benutzer das Experiment optimieren, beginnend mit einer hohen Konzentration (empfohlen 20 μM) für jeden Partner, um festzustellen, ob die Affinität der Komponenten innerhalb des Konzentrationsbereichs liegt, der durch die vorgestellte Methode aufrechterhalten wird (d. h. es bilden sich Komplexe). Die Optimierung kann auch die Wahl anderer Pufferbedingungen beinhalten, um die Wechselwirkungen zu fördern, oder die Titration einer der Wechselwirkungskomponenten, um das richtige Mischungsverhältnis zu bestimmen. Sobald diese bestimmt sind, ist es möglich, Konzentrationen und Durchflüsse zu optimieren, um optimale Bedingungen für die Studie und die Methode zu schaffen, z. B. die Konzentrationen zu verringern, um eine bessere Peakauflösung zu ermöglichen.
Zweitens, um dieses Experiment erfolgreich zu replizieren, sollten Verunreinigungen minimiert werden. Zu den häufigsten Quellen von Verunreinigungen, von denen bekannt ist, dass sie sich negativ auf Massenphotometriemessungen auswirken, gehören andere Proteine oder zelluläre Trümmer, die nach der Reinigung zurückbleiben, ungefilterte Puffer, mizellenbildende Detergenzien (wenn sie in zu hoher Konzentration vorhanden sind) und Puffer, die hohe Konzentrationen von Salz, Glycerin oder anderen Komponenten enthalten. Wie im obigen Protokoll beschrieben, sollten Blasen im Mikrofluidiksystem entfernt werden. Blasen können sich im Schlauchsystem bilden oder wenn Proben eine hohe Oberflächenspannung aufweisen und zur Schaumbildung neigen. Auch im Immersionsöl können sich Blasen bilden, die am Fokusring erkennbar sind (Abbildung 3). Wenn Blasen mit den im Protokoll beschriebenen Schritten nicht entfernt werden können, besteht eine andere Lösung darin, die Probe mit einem Exsikkator und einer Vakuumpumpe zu entgasen und die Probe einige Minuten lang unter reduziertem Druck zu belassen. Das Vortexen oder Schütteln hochkonzentrierter Proteinlösungen wird nicht empfohlen, da diese Maßnahmen die Blasenbildung fördern können.
Während hier die Messung einer spezifischen Protein-Protein-Interaktion demonstriert wird, kann das gleiche Protokoll ohne signifikante Modifikation auf andere Protein-Protein-Interaktionssysteme angewendet werden. Eine weitere zukünftige Richtung dieses Protokolls wäre es, die Messungen zur Berechnung von KD-Werten für die identifizierten Komplexe zu verwenden, wie dies an anderer Stelle im Rahmen der Massenphotometrie beschrieben wurde 5,7. Während in den vorangegangenen Studien Daten aus Experimenten mit manueller Verdünnung und stärkeren Wechselwirkungen verwendet wurden, könnte das Analyseprinzip in diesem Zusammenhang leicht angewendet werden – vorausgesetzt, es werden weitere Verbesserungen in der mikrofluidischen Vorrichtung implementiert (z. B. erhöhte Genauigkeit des Durchflusssensors und Pumpenstabilität).
Über Protein-Protein-Wechselwirkungen hinaus wird es wahrscheinlich breitere Anwendungen für die kombinierte Massenphotometrie und die Schnellverdünnungs-Mikrofluidik geben. Die Massenphotometrie kann zur Beurteilung der Reinheit, Aggregation und Homogenität von Proben verwendet werden18,19; Untersuchung der Proteinoligomerisierung20, der makromolekularen Assemblierung21 oder der Polymerisation22; und in anderen Bereichen. Die massenphotometrische Analyse geht auch über Proteine hinaus. Es wurde verwendet, um Wechselwirkungen zwischen Nukleinsäuren und Proteinen23, Viruspartikeln24 und Nanopartikeln25 zu untersuchen. Dieses Protokoll beschreibt somit eine wichtige Anwendung eines kombinierten Massenphotometrie-Mikrofluidik-Systems – es ermöglicht die direkte Messung schwacher Protein-Protein-Wechselwirkungen auf der Ebene einzelner Moleküle und Komplexe. Der Wert der vorliegenden Anwendung ist hoch, da sie die Möglichkeit eröffnet, Wechselwirkungen, die im Allgemeinen schwer zu untersuchen waren, einfach zu charakterisieren – mit Relevanz in kritischen therapeutischen Bereichen. Dieser kombinierte Ansatz könnte auch als Grundlage für ein breiteres Spektrum von Untersuchungen für Proben mit Konzentrationen bis zu einem Dutzend Mikromolaren dienen.
