To overcome the limitations of detecting ssDNA in G1, we utilized RPA2, which enhances both the specificity and intensity of ssDNA foci detection35. To achieve precise cell synchronization, we used RPE1 cells that can be efficiently serum-starved and synchronized into G0 phase. They can then be induced to re-enter the cell cycle by the addition of serum following serum deprivation. To confirm the synchronization efficiency, we labeled the cells with EdU and their DNA content with propidium-iodide. We further gathered qualitative and quantitative results via flow cytometry (Supplementary Figure S1A). The dot-plots show that after 72 h of serum starvation, ~98% of the cells are in G0 phase. Following the addition of serum-containing media for 6 h, cells reenter the cell cycle (as seen by the increase in p27 levels in Figure 1A), having ~97% of cells in G1, while only having <1% cells in S phase, <2% cells in G2 phase (Supplementary Figure S1A). After 20-28 h of addition of serum to the cells, they gradually pass through S phase, as shown by the flow cytometry plots (Supplementary Figure S1A). This cell synchronization protocol gives a ~97% pure G1 population (6 h post serum addition following 72 h of serum starvation). To further validate the synchronization efficiency, we compared the expression of cell cycle markers following serum release using western blotting (Figure 1A and Supplementary Figure S1B) and in parallel, performed an EdU incorporation assay to visualize DNA replication. The EdU staining also highlights synchronization efficiency and the lack of DNA replication in G1 phase (Figure 1B,C).
Conventional methods to detect ssDNA in mammalian cells rely on the detection of BrdU in ssDNA. Figure 2A demonstrates that upon H2O2 and neocarzinostatin (NCS) treatment, the BrdU foci were detectable only in S phase cells, while no ssDNA foci were detectable in non-S phase cells. The BrdU antibody staining also showed a noticeable nucleolar background staining that could be detected in all the nuclei, independent of the cell cycle stage or treatments applied. Using the EdU click protocol described here, we could not detect co-localizing EdU and BrdU foci, which is evident in the untreated samples of Figure 2A. To completely rule out any BrdU signal that arose from cross-reactivity, we avoided EdU labeling and rather used cyclin A2 as an S-G2 marker. However, cyclin A2 staining did not allow CSK pre-extraction, and under this condition, we did not see any BrdU foci, even after genotoxic stress (Supplementary Figure S2A). This highlights the fact that CSK pre-extraction is necessary for anti-BrdU-based ssDNA staining. As a control, we tested BrdU antibody staining under denaturing conditions. This opens the DNA to expose the incorporated BrdU, which reveals that BrdU was uniformly incorporated (Supplementary Figure S2B).
In contrast, RPA2 staining shows NCS- and H2O2-dependent foci formation not only in the S phase but also in other cell cycle phases (Figure 2B). As a control, we also treated the cells with HU, which only causes ssDNA accumulation in cells undergoing replication. As expected, we only detected a signal increase upon HU treatment with the RPA2 antibody in EdU-positive cells, highlighting the specificity of this approach. The RPA2 antibody can also detect naturally occurring ssDNA formation during replication in the absence of exogenous genotoxic stress (Figure 2B). The highly sensitive nature of the RPA2 antibody prompted us to try to utilize it in the G1 phase where conventional BrdU staining failed to detect any signal upon genotoxic stress (Supplementary Figure S2C). Figure 3A shows that the formation of ssDNA foci upon H2O2 treatment was detectable when using an anti-RPA2 antibody, even in G1. There was a significant increase in the number of RPA2 foci in these nuclei upon H2O2 treatment (Figure 3B). These foci were specific to RPA2 as silencing of RPA2 abolished the IF signal (Figure 3A,B). Figure 3C and Supplementary Figure S1C show the efficiency of RPA2 silencing in these cells. Compared to conventional methods, RPA2-based detection of ssDNA is highly sensitive, and its application can therefore be extended to G1 phase cells.
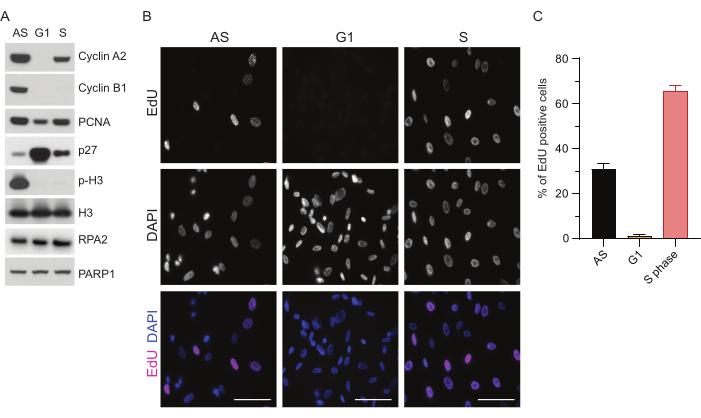
Figure 1: Synchronization efficiency of RPE1 cells following serum starvation. (A) Immunoblots show indicated protein levels in asynchronous, G1, and S phase synchronized RPE1 cells. (B) Representative images show asynchronous, G1, and S phase synchronized RPE1 cells that were exposed to 10 µM EdU for 30 min before fixation and visualized by Click-IT reaction. DAPI was used to counterstain nuclear DNA. Scale bars = 50 µm. (C) Graph shows percentage of EdU-positive cells over the total cell population assessed by DAPI. The error bar represents standard error of mean, and the analyzed numbers of nuclei were the following: AS n = 219, G1 n = 630, S n = 437. Abbreviations: RPE1 = hTERT-immortalized retinal pigment epithelial cells; AS = asynchronous; EdU = 5-ethynyl-2'-deoxyuridine; DAPI = 4',6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
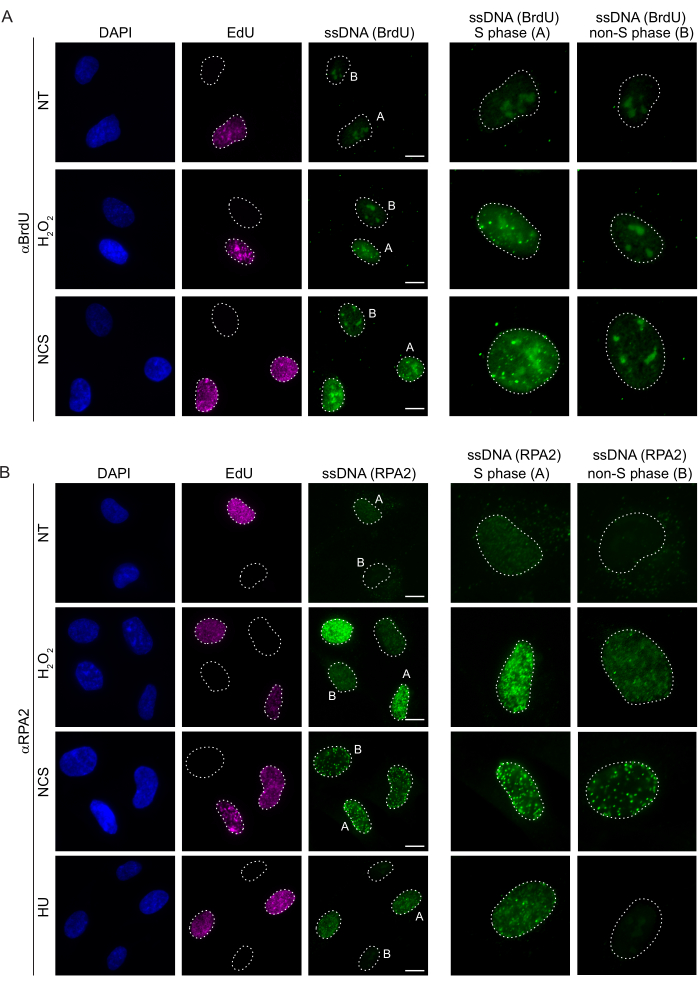
Figure 2: ssDNA detection with either BrdU antibody or RPA2 antibody upon DNA damage. (A) Representative images illustrate ssDNA foci using αBrdU (green), S phase cells are highlighted by EdU (purple), and DAPI was used to counterstain nuclear DNA (blue). RPE1 cells were kept in 10 µM BrdU for 48 h prior to any additional treatment. After 48 h, cells were pulsed with 10 µM EdU for 30 min followed by treatment of H2O2 (250 μM) for 1 h or Neocarzinostatin (0.5 μg/mL) for 4 h. Cells were fixed after CSK pre-extraction. A white dashed line denotes the border of each nucleus. Scale bar = 5 µm. Panels on the right are enlarged images of the indicated S phase or non-S phase nuclei. (B) Representative images illustrate ssDNA foci using αRPA2 antibody (green). S phase cells are highlighted by EdU (purple), and DAPI was used to counterstain nuclear DNA (blue). RPE1 cells were pulsed with 10 µM EdU for 30 min followed by either 1 h H2O2 (250 μM), 4 h of Hydroxyurea (2 mM), or 4 h of NCS (0.5 μg/mL). Cells were fixed after CSK pre-extraction. A white dashed line denotes the border of each nucleus. Scale bar = 10 µm. Panels on the right are enlarged images of the indicated S phase or non-S phase nuclei. Abbreviations: ssDNA = single-stranded DNA; BrdU = 5-bromo-2'-deoxyuridine; DAPI = 4',6-diamidino-2-phenylindole; RPE1 = hTERT-immortalized retinal pigment epithelial cells; EdU = 5-ethynyl-2'-deoxyuridine; NCS = Neocarzinostatin; HU = hydroxyurea. Please click here to view a larger version of this figure.
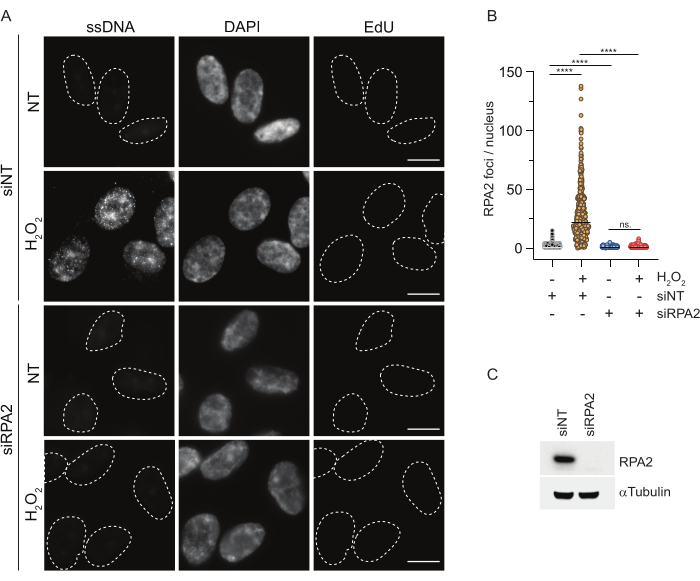
Figure 3: Detection of ssDNA foci in G1 phase using RPA2 antibody. (A) RPE1 cells were transfected with either siRNAs targeting RPA2 or a non-targeting siRNA control, and subsequently synchronized in G1 and pulse-labeled with 10 µM EdU for 30 min before treating them with H2O2 (250 μM) for 1 h where indicated. DAPI was used to counterstain nuclear DNA. Cells were fixed after CSK pre-extraction. A white dashed line denotes the border of each nucleus. Scale bar = 5 µm. (B) The measurements for the number of RPA2 foci/nucleus were carried out from two independent experiments. Only EdU-negative cells were considered during the analysis. Lines represent the mean value on the plots. Non-parametric ANOVA test (Kruskal-Wallis) was performed for statistical analysis. The stars indicate P < 0.0001. The analyzed number of nuclei were the following: siNT no H2O2 n = 513, siNT H2O2 n = 603, siRPA2 no H2O2 n = 266, siRPA2 H2O2 n = 536. (C) The efficiency of the siRNA knockdown is shown in the immunoblotting. Abbreviations: siNT = non-targeting siRNA control; BrdU = 5-bromo-2'-deoxyuridine; DAPI = 4',6-diamidino-2-phenylindole; RPE1 = hTERT-immortalized retinal pigment epithelial cells; EdU = 5-ethynyl-2'-deoxyuridine. Please click here to view a larger version of this figure.

Figure 4: Quantification of ssDNA foci using Fiji. Detailed steps in Fiji showing how to assess RPA2 foci numbers in the nucleus. (A–E) The creation of a nuclear mask using the DAPI channel. (F–H) Thresholding to identify individual nuclear ssDNA foci from the background signal. Abbreviations: ssDNA = single-stranded DNA; DAPI = 4',6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
| Cytoskeletal (CSK) buffer | |
| PIPES pH 7.0 | 10 mM |
| NaCl | 100 mM |
| EDTA pH 8 | 1 mM |
| MgCl2 | 3 mM |
| D-sucrose | 300 mM |
| Triton X-100 | 0.20% |
| Phosphatase inhibitor cocktail | 1 tablet per 10 mL |
| Protease inhibitor cocktail | 1 tablet per 10 mL |
| diluted in ddH2O | n/a |
| Washing buffer | |
| Triton X-100 | 0.05% |
| diluted in PBS | n/a |
| Permeabilization buffer | |
| Triton X-100 | 0.50% |
| diluted in PBS | n/a |
| Fixation solution | |
| Paraformaldehyde | 3.60% |
| Triton X-100 | 0.05% |
| diluted in PBS | n/a |
| Blocking buffer | |
| Bovine serum albumin (BSA) | 5% |
| Triton X-100 | 0.10% |
| diluted in PBS | n/a |
| Click-iT Plus reaction cocktail | |
| 1x Click-iT reaction buffer | 435 mL |
| Alexa Fluor PCA solution | 5 mL |
| CuSO4-copper protectant premix | 10 mL |
| 1x Click-iT buffer additive | 50 mL |
| Total volume | 500 mL |
Table 1: Composition of the buffers used in this protocol.
Supplementary Figure S1. (A) RPE1 cells were synchronized to G0 phase using serum starvation for 72 h and subsequently released into different cell cycle phases by reintroducing serum. Dot plots show cells in G0/G1, S, or G2/M phases, where hours indicate the time after the re-addition of serum following serum starvation. Graph on the right shows the percentage of G0/G1, S, and G2/M cells in each condition. FACS analysis was carried out using a commercially available cell proliferation kit using EdU and propidium iodide according to the manufacturer's recommendations. (B) Uncropped western blot scans for Figure 1. Numbers show molecular weight markers in kDa. PARP1 was used as a loading control and loaded on the gel that was also developed against CCNA2, p27 (further stripped for PCNA), and pH3 (S10) (further stripped for H3) by cutting up the membrane. CCNB1 and RPA2 were loaded onto a separate gel, using the same amount of protein lysate to ensure comparability. (C) Uncropped western blot scans for Figure 3. Numbers show molecular weight markers in kDa. Abbreviation: EdU = 5-ethynyl-2'-deoxyuridine. Please click here to download this File.
Supplementary Figure S2: (A) Representative images illustrate ssDNA foci using BrdU antibody (green); S phase cells are highlighted by cyclin A2 (red); and DAPI was used to counterstain nuclear DNA (blue). RPE1 cells were kept in 10 µM BrdU for 48 h prior to further treatment. After 48 h, cells were treated with H2O2 (250 μM) for 1 h or Neocarzinostatin (0.5 µg/mL) for 4 h before fixation. A white dashed line denotes the border of each nucleus. Scale bar = 5 µm. (B) BrdU staining of RPE1 cells with and without denaturing condition. Asynchronous RPE1 cells were pre-treated with 10 µM BrdU for 48 h. Scale bar = 10 µm. (C) The measurements for the number of BrdU foci/nucleus were carried out from two independent experiments in G1 synchronized RPE1 cells. Only EdU-negative cells were considered during the analysis. Lines represent the mean value on the plots. Non-parametric ANOVA test (Kruskal-Wallis) was performed for statistical analysis. The 'ns' indicates non-significant difference. The analyzed number of nuclei were the following: NT n = 52, NCS n = 105, H2O2 n = 82. Abbreviations: siNT = non-targeting siRNA control; BrdU = 5-bromo-2'-deoxyuridine; DAPI = 4',6-diamidino-2-phenylindole; RPE1 = hTERT-immortalized retinal pigment epithelial cells; NCS = Neocarzinostatin. Please click here to download this File.
Supplementary Video S1: Screen recording of Fiji-based RPA2 foci analysis. Please click here to download this File.
