يتكون Transcriptome من تعبيرات جميع الجينات في العينة ويمكن توصيفه بواسطة تقنيات عالية الإنتاجية مثل microarray و RNA-seq1. تسمى مستويات التعبير لجين واحد في مجموعة البيانات ميزة النسخ ، والتمثيل التفاضلي لميزة النسخ بين النمط الظاهري والمجموعات الضابطة يعرف هذا الجين كعلامة حيوية لهذا النمط الظاهري 2,3. تم استخدام المؤشرات الحيوية النسخية على نطاق واسع في التحقيقات في تشخيص المرض4 ، والآلية البيولوجية5 ، وتحليل البقاء على قيد الحياة6،7 ، إلخ.
تحمل أنماط نشاط الجينات في الأنسجة السليمة معلومات مهمة عن حياة 8,9. تقدم هذه الأنماط رؤى لا تقدر بثمن وتعمل كمراجع مثالية لفهم المسارات التنموية المعقدة للاضطرابات الحميدة10,11 والأمراض الفتاكة12. تتفاعل الجينات مع بعضها البعض ، وتمثل النسخ مستويات التعبير النهائية بعد تفاعلاتها المعقدة. تتم صياغة هذه الأنماط مثل شبكة تنظيم النسخ13 وشبكة التمثيل الغذائي14 ، إلخ. يمكن تنظيم تعبيرات الحمض النووي الريبي المرسال (mRNAs) نسخيا بواسطة عوامل النسخ (TFs) والحمض النووي الريبي الطويل غير المشفر بين الجينات (lincRNAs)15،16،17. تجاهل تحليل التعبير التفاضلي التقليدي مثل هذه التفاعلات الجينية المعقدة مع افتراض الاستقلال بين الميزات18,19.
تظهر التطورات الحديثة في الشبكات العصبية للرسم البياني (GNNs) إمكانات غير عادية في استخراج المعلومات المهمة من البيانات المستندة إلى OMIC لدراسات السرطان20 ، على سبيل المثال ، تحديد وحدات التعبير المشترك21. القدرة الفطرية ل GNNs تجعلها مثالية لنمذجة العلاقات المعقدة والتبعيات بين الجينات22,23.
غالبا ما تركز الدراسات الطبية الحيوية على التنبؤ بدقة بالنمط الظاهري مقابل المجموعة الضابطة. عادة ما تصاغ هذه المهام كتصنيفات ثنائية24،25،26. هنا ، عادة ما يتم ترميز تسميات الفئتين على أنها 1 و 0 ، صواب وخطأ ، أو حتى موجب وسالب27.
تهدف هذه الدراسة إلى توفير بروتوكول سهل الاستخدام لتوليد عرض تنظيم النسخ (mqTrans) لمجموعة بيانات النسخ بناء على النموذج المرجعي لشبكة انتباه الرسم البياني (GAT) المدرب مسبقا. تم استخدام إطار عمل GAT متعدد المهام من عمل منشور سابقا26 لتحويل ميزات النسخ إلى ميزات mqTrans. تم استخدام مجموعة بيانات كبيرة من النسخ الصحية من جامعة كاليفورنيا ، سانتا كروز (UCSC) منصة Xena28 للتدريب المسبق للنموذج المرجعي (HealthModel) ، والذي يقيس كميا لوائح النسخ من العوامل التنظيمية (TFs و lincRNAs) إلى mRNAs المستهدفة. يمكن استخدام عرض mqTrans الذي تم إنشاؤه لبناء نماذج التنبؤ واكتشاف المؤشرات الحيوية المظلمة. يستخدم هذا البروتوكول مجموعة بيانات مرضى سرطان القولون الغدي (COAD) من قاعدة بيانات أطلس جينوم السرطان (TCGA)29 كمثال توضيحي. في هذا السياق ، يتم تصنيف المرضى في المرحلتين الأولى أو الثانية على أنهم عينات سلبية ، بينما يعتبر المرضى في المرحلتين الثالثة أو الرابعة عينات إيجابية. كما تتم مقارنة توزيعات المؤشرات الحيوية الداكنة والتقليدية عبر 26 نوعا من أنواع السرطان TCGA.
وصف خط أنابيب HealthModel
تستند المنهجية المستخدمة في هذا البروتوكول إلى الإطار26 المنشور سابقا ، كما هو موضح في الشكل 1. للبدء، يطلب من المستخدمين إعداد مجموعة بيانات الإدخال، وإدخالها في خط أنابيب HealthModel المقترح، والحصول على ميزات mqTrans. وترد تعليمات مفصلة لإعداد البيانات في القسم 2 من قسم البروتوكول. بعد ذلك، يتوفر للمستخدمين خيار دمج ميزات mqTrans مع ميزات النسخ الأصلية أو متابعة ميزات mqTrans التي تم إنشاؤها فقط. ثم تخضع مجموعة البيانات المنتجة لعملية اختيار الميزات ، حيث يتمتع المستخدمون بالمرونة في اختيار القيمة المفضلة لديهم ل k في التحقق المتقاطع k-fold للتصنيف. مقياس التقييم الأساسي المستخدم في هذا البروتوكول هو الدقة.
يصنف HealthModel26 ميزات النسخ إلى ثلاث مجموعات متميزة: TF (عامل النسخ) ، lincRNA (الحمض النووي الريبي الطويل غير المشفر بين الجينات) ، و mRNA (الحمض النووي الريبي المرسال). يتم تعريف ميزات TF بناء على التعليقات التوضيحية المتوفرة في أطلس البروتين البشري30,31. يستخدم هذا العمل التعليقات التوضيحية ل lincRNAs من مجموعة بياناتGTEx 32. تعتبر الجينات التي تنتمي إلى مسارات المستوى الثالث في قاعدة بيانات KEGG33 من ميزات mRNA. تجدر الإشارة إلى أنه إذا أظهرت ميزة mRNA أدوارا تنظيمية للجين المستهدف كما هو موثق في قاعدة بيانات TRRUST34 ، إعادة تصنيفها في فئة TF.
يقوم هذا البروتوكول أيضا بإنشاء ملفين نموذجيين يدويا لمعرفات الجينات للعوامل التنظيمية (regulatory_geneIDs.csv) و mRNA المستهدف (target_geneIDs.csv). يتم حساب مصفوفة المسافة الزوجية بين السمات التنظيمية (TFs و lincRNAs) بواسطة معاملات ارتباط بيرسون ويتم تجميعها بواسطة أداة تحليل شبكة التعبير المشترك للجين المرجح (WGCNA) 36 (adjacent_matrix.csv). يمكن للمستخدمين استخدام خط أنابيب HealthModel مباشرة مع ملفات التكوين النموذجية هذه لإنشاء عرض mqTrans لمجموعة بيانات نسخية.
التفاصيل الفنية لنموذج الصحة
يمثل HealthModel العلاقات المعقدة بين TFs و lincRNAs كرسم بياني ، مع ميزات الإدخال التي تعمل كرؤوس يشار إليها بواسطة V ومصفوفة حافة بين الرؤوس تسمى E. تتميز كل عينة بميزات تنظيمية K ، يرمز لها ب VK ×1. على وجه التحديد ، شملت مجموعة البيانات 425 TFs و 375 lincRNAs ، مما أدى إلى أبعاد عينة من K = 425 + 375 = 800. لإنشاء مصفوفة الحافة E ، استخدم هذا العمل الأداة الشعبية WGCNA35. يتم تحديد الوزن الزوجي الذي يربط رأسين ممثلين ك 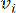 و
و 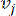 ، بواسطة معامل ارتباط بيرسون. تعرض شبكة تنظيم الجينات طوبولوجيا خالية من المقياس36 ، تتميز بوجود جينات محورية ذات أدوار وظيفية محورية. نحسب الارتباط بين ميزتين أو رأسين ، و ، باستخدام مقياس التداخل الطوبولوجي (TOM) على النحو التالي:
، بواسطة معامل ارتباط بيرسون. تعرض شبكة تنظيم الجينات طوبولوجيا خالية من المقياس36 ، تتميز بوجود جينات محورية ذات أدوار وظيفية محورية. نحسب الارتباط بين ميزتين أو رأسين ، و ، باستخدام مقياس التداخل الطوبولوجي (TOM) على النحو التالي:
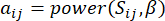 (1)
(1)
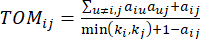 (2)
(2)
يتم حساب β العتبة اللينة باستخدام وظيفة “عتبة pickSoft” من حزمة WGCNA. يتم تطبيق دالة أس القدرة aij ، حيث 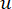 تمثل جينا باستثناء i و j ، وتمثل
تمثل جينا باستثناء i و j ، وتمثل 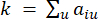 اتصال الرأس. يقوم WGCNA بتجميع ملفات تعريف التعبير لميزات النسخ في وحدات متعددة باستخدام مقياس الاختلاف المستخدم بشكل شائع (
اتصال الرأس. يقوم WGCNA بتجميع ملفات تعريف التعبير لميزات النسخ في وحدات متعددة باستخدام مقياس الاختلاف المستخدم بشكل شائع (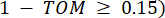 37.
37.
تم تصميم إطار عمل HealthModel في الأصل كبنية تعليمية متعددة المهام26. يستخدم هذا البروتوكول فقط مهمة التدريب المسبق النموذجية لبناء طريقة عرض mqTrans النسخية. يمكن للمستخدم اختيار زيادة تحسين HealthModel المدرب مسبقا ضمن شبكة انتباه الرسم البياني متعدد المهام مع عينات نسخ إضافية خاصة بالمهمة.
التفاصيل الفنية لاختيار الميزة وتصنيفها
ينفذ تجمع تحديد المعالم أحد عشر خوارزمية لتحديد المعالم (FS). من بينها ، ثلاثة خوارزميات FS قائمة على المرشح: اختيار أفضل ميزات K باستخدام معامل المعلومات الأقصى (SK_mic) ، واختيار ميزات K بناء على FPR ل MIC (SK_fpr) ، واختيار ميزات K ذات أعلى معدل اكتشاف خاطئ ل MIC (SK_fdr). بالإضافة إلى ذلك ، تقوم ثلاث خوارزميات FS قائمة على الأشجار بتقييم الميزات الفردية باستخدام شجرة القرار مع مؤشر Gini (DT_gini) ، وأشجار القرار المعززة التكيفية (AdaBoost) ، والغابة العشوائية (RF_fs). يشتمل التجمع أيضا على طريقتين للتغليف: إزالة الميزة العودية باستخدام مصنف متجه الدعم الخطي (RFE_SVC) والتخلص من الميزة العودية باستخدام مصنف الانحدار اللوجستي (RFE_LR). أخيرا ، يتم تضمين خوارزميتين للتضمين: مصنف SVC الخطي مع قيم أهمية ميزة L1 الأعلى مرتبة (lSVC_L1) ومصنف الانحدار اللوجستي مع قيم أهمية ميزة L1 الأعلى مرتبة (LR_L1).
يستخدم تجمع المصنف سبعة مصنفات مختلفة لبناء نماذج تصنيف. تشتمل هذه المصنفات على آلة متجه الدعم الخطي (SVC) ، و Gaussian Naïve Bayes (GNB) ، ومصنف الانحدار اللوجستي (LR) ، وأقرب جار k ، مع تعيين k على 5 افتراضيا (KNN) ، و XGBoost ، والغابة العشوائية (RF) ، وشجرة القرار (DT).
يمكن تعيين التقسيم العشوائي لمجموعة البيانات في القطار: مجموعات فرعية للاختبار في سطر الأوامر. يستخدم المثال الموضح نسبة القطار: اختبار = 8: 2.
