トランスクリプトームは、サンプル中のすべての遺伝子の発現で構成されており、マイクロアレイやRNA-seq1などのハイスループット技術によってプロファイリングすることができます。データセット内の1つの遺伝子の発現レベルはトランスクリプトーム特徴と呼ばれ、表現型と対照群の間でトランスクリプトームの特徴が異なって表現されることで、この遺伝子はこの表現型のバイオマーカーとして定義されます2,3。トランスクリプトームバイオマーカーは、疾患診断4、生物学的メカニズム5、生存解析6,7などの研究に広く利用されています。
健康な組織の遺伝子活性パターンは、生命に関する重要な情報を運びます8,9。これらのパターンは、非常に貴重な洞察を提供し、良性疾患10,11および致死性疾患12の複雑な発達の軌跡を理解するための理想的な参考資料として機能します。遺伝子は互いに相互作用し、トランスクリプトームは複雑な相互作用の後の最終的な発現レベルを表します。このようなパターンは、転写調節ネットワーク13および代謝ネットワーク14等として定式化される。メッセンジャーRNA(mRNA)の発現は、転写因子(TF)と長鎖遺伝子間ノンコーディングRNA(lincRNA)によって転写制御されている15,16,17。従来の差次的発現解析では、このような複雑な遺伝子相互作用は無視され、特徴間の独立性を仮定していた18,19。
グラフニューラルネットワーク(GNN)の最近の進歩は、がん研究20、例えば共発現モジュールの同定21など、OMICベースのデータから重要な情報を抽出する上で並外れた可能性を示しています。GNNの生来の能力は、遺伝子間の複雑な関係と依存関係をモデル化するのに理想的です22,23。
生物医学研究では、対照群に対する表現型を正確に予測することに重点が置かれることがよくあります。このようなタスクは、一般に二項分類24、25、26として定式化されます。ここで、2 つのクラス ラベルは通常、1 と 0、true と false、または正と負の27 としてエンコードされます。
この研究は、事前学習済みのグラフアテンションネットワーク(GAT)参照モデルに基づいて、トランスクリプトームデータセットの転写制御(mqTrans)ビューを生成するための使いやすいプロトコルを提供することを目的としています。以前に発表された研究26 のマルチタスクGATフレームワークを使用して、トランスクリプトームの特徴をmqTransの特徴に変換しました。カリフォルニア大学サンタクルーズ校(UCSC)のXenaプラットフォーム28 の健康なトランスクリプトームの大規模なデータセットを使用して、制御因子(TFおよびlincRNA)から標的mRNAまでの転写制御を定量的に測定する参照モデル(HealthModel)を事前トレーニングしました。生成されたmqTransビューは、予測モデルの構築やダークバイオマーカーの検出に使用できます。このプロトコルは、例示的な例として、がんゲノムアトラス(TCGA)データベース29 からの結腸腺癌(COAD)患者データセットを利用します。これに関連して、ステージIまたはIIの患者は陰性サンプルに分類され、ステージIIIまたはIVの患者は陽性サンプルと見なされます。また、26種類のTCGAがんにおけるダークバイオマーカーと従来のバイオマーカーの分布も比較しています。
HealthModel パイプラインの説明
このプロトコルで採用されている方法論は、図1に概説されているように、以前に公開されたフレームワーク26に基づいています。まず、ユーザーは入力データセットを準備し、提案されたHealthModelパイプラインにフィードし、mqTrans特徴を取得する必要があります。詳細なデータ準備手順は、プロトコルセクションのセクション2に記載されています。その後、ユーザーは、mqTransの特徴を元のトランスクリプトームの特徴と組み合わせるか、生成されたmqTransの特徴のみを続行するかを選択できます。生成されたデータセットは、特徴選択プロセスにかけられ、ユーザーは分類のためのk分割交差検証でkの優先値を柔軟に選択できます。このプロトコルで使用される主な評価指標は精度です。
HealthModel26は、トランスクリプトームの特徴をTF(転写因子)、lincRNA(長鎖遺伝子間ノンコーディングRNA)、mRNA(メッセンジャーRNA)の3つのグループに分類しています。TFの特徴は、Human ProteinAtlas30,31で利用可能なアノテーションに基づいて定義されています。この研究は、GTExデータセット32からのlincRNAのアノテーションを利用しています。KEGGデータベース33における第3レベルの経路に属する遺伝子は、mRNAの特徴と見なされる。TRRUSTデータベース34に記録されているように、mRNAの特徴が標的遺伝子に対する調節的役割を示す場合、それはTFクラスに再分類されることは注目に値する。
また、このプロトコルでは、規定因子(regulatory_geneIDs.csv)と標的mRNA(target_geneIDs.csv)の遺伝子IDの2つのサンプルファイルを手動で生成します。調節特性(TFおよびlincRNA)間のペアワイズ距離マトリックスは、ピアソン相関係数によって計算され、一般的なツール加重遺伝子共発現ネットワーク解析(WGCNA)36 (adjacent_matrix.csv)によってクラスタリングされます。ユーザーは、HealthModel パイプラインをこれらのサンプル構成ファイルと共に直接利用して、トランスクリプトーム データセットの mqTrans ビューを生成できます。
HealthModelの技術的詳細
HealthModel は、TF と lincRNA の間の複雑な関係をグラフとして表し、入力特徴は V で示される頂点として機能し、頂点間エッジ行列は E として示されます。各サンプルは、VK×1として表されるK個の調節特徴によって特徴付けられます。具体的には、データセットには 425 の TF と 375 の lincRNA が含まれ、サンプルの次元は K = 425 + 375 = 800 でした。エッジ行列 E を確立するために、この作業では一般的なツール WGCNA35 を使用しました。と 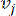 で表される
で表される 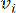 2 つの頂点をつなぐペアワイズ重みは、ピアソン相関係数によって決定されます。遺伝子調節ネットワークは、極めて重要な機能的役割を有するハブ遺伝子の存在を特徴とするスケールフリートポロジー36を呈する。2 つの特徴または頂点 と の間の相関関係は、トポロジカル オーバーラップ メジャー (TOM) を使用して次のように計算します。
2 つの頂点をつなぐペアワイズ重みは、ピアソン相関係数によって決定されます。遺伝子調節ネットワークは、極めて重要な機能的役割を有するハブ遺伝子の存在を特徴とするスケールフリートポロジー36を呈する。2 つの特徴または頂点 と の間の相関関係は、トポロジカル オーバーラップ メジャー (TOM) を使用して次のように計算します。
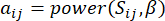 (1)
(1)
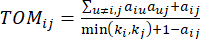 (2)
(2)
ソフトしきい値βは、WGCNA パッケージの ‘pickSoft Threshold’ 関数を使用して計算されます。べき乗指数関数 aij が適用され、ここで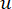 、 は i と j を除いた遺伝子を表し、
、 は i と j を除いた遺伝子を表し、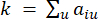 頂点接続性を表します。WGCNAは、トランスクリプトームの特徴の発現プロファイルを、一般的に採用されている非類似度尺度
頂点接続性を表します。WGCNAは、トランスクリプトームの特徴の発現プロファイルを、一般的に採用されている非類似度尺度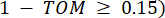 を用いて複数のモジュールにクラスタリングする(37.
を用いて複数のモジュールにクラスタリングする(37.
HealthModelフレームワークは、もともとマルチタスク学習アーキテクチャ26として設計されました。このプロトコルは、トランスクリプトーム mqTrans ビューの構築にモデルの事前トレーニング タスクのみを利用します。ユーザは、追加のタスク特異的トランスクリプトームサンプルを用いて、マルチタスクグラフアテンションネットワークの下で事前訓練されたHealthModelをさらに洗練させることを選択することができる。
機能の選択と分類に関する技術的な詳細
特徴選択プールには、11 個の特徴選択 (FS) アルゴリズムが実装されています。その中には、最大情報量係数(SK_mic)を使用したK個の最良の特徴量の選択、MICのFPRに基づくK個の特徴量の選択(SK_fpr)、MICの偽発見率が最も高いK個の特徴量の選択(SK_fdr)の3つがフィルタベースのFSアルゴリズムです。さらに、3つのツリーベースのFSアルゴリズムは、ジニ指数(DT_gini)、適応型ブースト決定木(AdaBoost)、およびランダムフォレスト(RF_fs)を備えた決定木を使用して個々の特徴を評価します。また、このプールには、線形サポートベクター分類器による再帰的特徴量消去法 (RFE_SVC) とロジスティック回帰分類器 (RFE_LR) による再帰的特徴量消去法の 2 つのラッパー手法も組み込まれています。最後に、最上位の L1 特徴重要度値 (lSVC_L1) を持つ線形 SVC 分類器と、最上位の L1 特徴重要度値 (LR_L1) を持つロジスティック回帰分類器の 2 つの埋め込みアルゴリズムが含まれています。
分類子プールでは、分類モデルを構築するために 7 つの異なる分類子が採用されています。これらの分類器は、線形サポート ベクター マシン (SVC)、ガウス単純ベイズ (GNB)、ロジスティック回帰分類器 (LR)、k 最近傍 (k は既定で 5 に設定されている) (KNN)、XGBoost、ランダム フォレスト (RF)、および決定木 (DT) で構成されます。
データセットをtrain:testサブセットにランダムに分割する方法は、コマンドラインで設定できます。この例では、train: test = 8:2 の比率を使用します。
