Transkriptom består av uttrykkene av alle genene i en prøve og kan profileres av høykapasitetsteknologier som mikroarray og RNA-seq1. Ekspresjonsnivåene til ett gen i et datasett kalles en transkriptomisk funksjon, og differensialrepresentasjonen av et transkriptomisk trekk mellom fenotype- og kontrollgruppene definerer dette genet som en biomarkør for denne fenotypen 2,3. Transkriptomiske biomarkører har blitt mye brukt i undersøkelsene av sykdomsdiagnose4, biologisk mekanisme5 og overlevelsesanalyse 6,7, etc.
Genaktivitetsmønstre i det friske vevet bærer viktig informasjon om livene 8,9. Disse mønstrene gir uvurderlig innsikt og fungerer som ideelle referanser for å forstå de komplekse utviklingsbanene til godartede lidelser 10,11 og dødelige sykdommer12. Gener interagerer med hverandre, og transkriptomer representerer de endelige uttrykksnivåene etter deres kompliserte interaksjoner. Slike mønstre er formulert som transkripsjonsreguleringsnettverk13 og metabolismenettverk14, etc. Uttrykkene av budbringer-RNA (mRNA) kan transkripsjonelt reguleres av transkripsjonsfaktorer (TF) og lange intergeniske ikke-kodende RNA (lincRNAer)15,16,17. Konvensjonell differensialekspresjonsanalyse ignorerte slike komplekse geninteraksjoner med antagelsen om uavhengighet mellom funksjoner 18,19.
Nylige fremskritt i grafnevrale nettverk (GNN) viser ekstraordinært potensial i å trekke ut viktig informasjon fra OMIC-baserte data for kreftstudier20, for eksempel å identifisere co-uttrykksmoduler21. Den medfødte kapasiteten til GNN gjør dem ideelle for modellering av de intrikate forholdene og avhengighetene mellom gener22,23.
Biomedisinske studier fokuserer ofte på nøyaktig å forutsi en fenotype mot kontrollgruppen. Slike oppgaver er vanligvis formulert som binære klassifikasjoner 24,25,26. Her er de to klasseetikettene vanligvis kodet som 1 og 0, sant og usant, eller til og med positivt og negativt27.
Denne studien hadde som mål å gi en brukervennlig protokoll for generering av transkripsjonsregulering (mqTrans) visning av et transkriptomdatasett basert på den forhåndstrente graf-oppmerksomhetsnettverket (GAT) referansemodell. Multitask GAT-rammeverket fra et tidligere publisert arbeid26 ble brukt til å transformere transkriptomiske funksjoner til mqTrans-funksjonene. Et stort datasett med friske transkriptomer fra University of California, Santa Cruz (UCSC) Xena-plattform28 ble brukt til å forhåndstrene referansemodellen (HealthModel), som kvantitativt målte transkripsjonsforskriftene fra regulatoriske faktorer (TF og lincRNA) til mål-mRNAene. Den genererte mqTrans-visningen kan brukes til å bygge prediksjonsmodeller og oppdage mørke biomarkører. Denne protokollen bruker pasientdatasettet for kolonadenokarsinom (COAD) fra The Cancer Genome Atlas (TCGA) database29 som et illustrerende eksempel. I denne sammenheng kategoriseres pasienter i stadium I eller II som negative prøver, mens de i stadium III eller IV regnes som positive prøver. Fordelingen av mørke og tradisjonelle biomarkører på tvers av de 26 TCGA-krefttypene sammenlignes også.
Beskrivelse av HealthModel-pipelinen
Metodikken som benyttes i denne protokollen er basert på det tidligere publiserte rammeverket26, som skissert i figur 1. For å starte, må brukerne klargjøre inndatasettet, mate det inn i den foreslåtte HealthModel-pipelinen og få mqTrans-funksjoner. Detaljerte instruksjoner for dataforberedelse er gitt i avsnitt 2 i protokollseksjonen. Deretter har brukerne muligheten til å kombinere mqTrans-funksjoner med de originale transkriptomiske funksjonene eller bare fortsette med de genererte mqTrans-funksjonene. Det produserte datasettet blir deretter utsatt for en funksjonsvalgsprosess, der brukerne har fleksibilitet til å velge sin foretrukne verdi for k i k-fold kryssvalidering for klassifisering. Den primære evalueringsmålingen som brukes i denne protokollen, er nøyaktighet.
HealthModel26 kategoriserer de transkriptomiske funksjonene i tre forskjellige grupper: TF (transkripsjonsfaktor), lincRNA (langt intergenisk ikke-kodende RNA) og mRNA (messenger RNA). TF-funksjonene er definert basert på merknadene som er tilgjengelige i Human Protein Atlas30,31. Dette arbeidet benytter merknadene til lincRNA fra GTEx-datasettet32. Gener som tilhører tredjenivåbanene i KEGG-databasen33 regnes som mRNA-egenskaper. Det er verdt å merke seg at hvis en mRNA-funksjon viser regulatoriske roller for et målgen som dokumentert i TRRUST-databasen34, blir den omklassifisert til TF-klassen.
Denne protokollen genererer også manuelt de to eksempelfilene for gen-IDene til regulatoriske faktorer (regulatory_geneIDs.csv) og mål-mRNA (target_geneIDs.csv). Den parvise avstandsmatrisen mellom regulatoriske trekk (TF og lincRNA) beregnes av Pearson-korrelasjonskoeffisientene og grupperes av den populære verktøyvektede genkoekspresjonsnettverksanalysen (WGCNA) 36 (adjacent_matrix.csv). Brukere kan bruke HealthModel-pipelinen direkte sammen med disse eksempelkonfigurasjonsfilene for å generere mqTrans-visningen av et transkriptomisk datasett.
Tekniske detaljer om HealthModel
HealthModel representerer de intrikate forholdene mellom TF og lincRNA som en graf, med inngangsfunksjonene som fungerer som hjørnene betegnet med V og en inter-toppunktkantmatrise betegnet som E. Hver prøve er preget av K-regulatoriske egenskaper, symbolisert som VK×1. Spesifikt omfattet datasettet 425 TF og 375 lincRNA, noe som resulterte i en prøvedimensjonalitet på K = 425 + 375 = 800. For å etablere kantmatrisen E brukte dette arbeidet det populære verktøyet WGCNA35. Den parvise vekten som forbinder to hjørner representert som 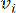 og
og 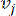 , bestemmes av Pearson-korrelasjonskoeffisienten. Det genregulerende nettverket utviser en skalafri topologi36, preget av tilstedeværelsen av navgener med sentrale funksjonelle roller. Vi beregner korrelasjonen mellom to funksjoner eller toppunkter, og , ved hjelp av det topologiske overlappingsmålet (TOM) som følger:
, bestemmes av Pearson-korrelasjonskoeffisienten. Det genregulerende nettverket utviser en skalafri topologi36, preget av tilstedeværelsen av navgener med sentrale funksjonelle roller. Vi beregner korrelasjonen mellom to funksjoner eller toppunkter, og , ved hjelp av det topologiske overlappingsmålet (TOM) som følger:
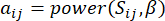 (1)
(1)
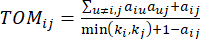 (2)
(2)
Den myke terskelen β beregnes ved hjelp av funksjonen “pickSoft Threshold” fra WGCNA-pakken. Potenseksponentialfunksjonen aij brukes, der 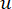 representerer et gen som ekskluderer i og j, og
representerer et gen som ekskluderer i og j, og 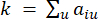 representerer toppunktforbindelsen. WGCNA grupperer uttrykksprofilene til de transkriptomiske trekkene i flere moduler ved hjelp av et vanlig ulikhetsmål (
representerer toppunktforbindelsen. WGCNA grupperer uttrykksprofilene til de transkriptomiske trekkene i flere moduler ved hjelp av et vanlig ulikhetsmål (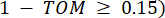 37.
37.
HealthModel-rammeverket ble opprinnelig designet som en multitask læringsarkitektur26. Denne protokollen benytter bare modellens fortreningsoppgave for konstruksjon av den transkriptomiske mqTrans-visningen. Brukeren kan velge å videreutvikle den forhåndstrente HealthModel under multitask graph attention network med flere oppgavespesifikke transkriptomiske prøver.
Tekniske detaljer om funksjonsvalg og klassifisering
Funksjonsutvalget implementerer elleve algoritmer for funksjonsvalg (FS). Blant dem er tre filterbaserte FS-algoritmer: velge K beste funksjoner ved hjelp av maksimal informasjonskoeffisient (SK_mic), velge K-funksjoner basert på FPR for MIC (SK_fpr), og velge K-funksjoner med den høyeste falske oppdagelsesfrekvensen for MIC (SK_fdr). I tillegg vurderer tre trebaserte FS-algoritmer individuelle funksjoner ved hjelp av et beslutningstre med Gini-indeksen (DT_gini), adaptive boosted decision trees (AdaBoost) og random forest (RF_fs). Bassenget inneholder også to wrapper-metoder: Rekursiv funksjonseliminering med lineær støttevektorklassifiserer (RFE_SVC) og eliminering av rekursive funksjoner med logistisk regresjonsklassifiserer (RFE_LR). Til slutt inkluderes to innebyggingsalgoritmer: lineær SVC-klassifiserer med de topprangerte L1-funksjonsviktighetsverdiene (lSVC_L1) og logistisk regresjonsklassifiserer med de topprangerte L1-funksjonsviktighetsverdiene (LR_L1).
Klassifiseringsutvalget bruker syv forskjellige klassifiserere for å bygge klassifiseringsmodeller. Disse klassifisererne omfatter lineær støttevektormaskin (SVC), Gaussian Naïve Bayes (GNB), logistisk regresjonsklassifiserer (LR), k-nærmeste nabo, med k satt til 5 som standard (KNN), XGBoost, tilfeldig skog (RF) og beslutningstre (DT).
Den tilfeldige delingen av datasettet i toget: testundergrupper kan settes i kommandolinjen. Det demonstrerte eksemplet bruker forholdet mellom tog: test = 8: 2.
