O transcriptoma consiste na expressão de todos os genes de uma amostra e pode ser perfilado por tecnologias de alto rendimento, como microarray e RNA-seq1. Os níveis de expressão de um gene em um conjunto de dados são chamados de característica transcriptômica, e a representação diferencial de uma característica transcriptômica entre os grupos fenótipo e controle define esse gene como um biomarcador desse fenótipo 2,3. Biomarcadores transcriptômicos têm sido extensivamente utilizados em investigações de diagnóstico dedoenças4, mecanismobiológico5, análise desobrevida6,7, etc.
Os padrões de atividade gênica nos tecidos sadios carregam informações cruciais sobre as vidas 8,9. Esses padrões oferecem insights inestimáveis e funcionam como referências ideais para a compreensão das complexas trajetórias de desenvolvimento das doençasbenignas10,11 eletais12. Os genes interagem entre si, e os transcriptomas representam os níveis finais de expressão após suas complicadas interações. Tais padrões são formulados como rede de regulação transcricional13 e rede de metabolismo14, etc. A expressão de RNAs mensageiros (mRNAs) pode ser regulada transcricionalmente por fatores de transcrição (FTs) e RNAs não codificadores intergênicos longos (lincRNAs)15,16,17. A análise convencional de expressão diferencial ignorou interações gênicas tão complexas com a suposição de independência inter-características18,19.
Avanços recentes em redes neurais de grafos (GNNs) demonstram um potencial extraordinário na extração de informações importantes de dados baseados em OMIC para estudos de câncer20, por exemplo, identificando módulos de co-expressão21. A capacidade inata dos GNNs os torna ideais para modelar as intrincadas relações e dependências entre genes22,23.
Estudos biomédicos geralmente se concentram em prever com precisão um fenótipo contra o grupo controle. Tais tarefas são comumente formuladas como classificações binárias 24,25,26. Aqui, os dois rótulos de classe são tipicamente codificados como 1 e 0, verdadeiro e falso, ou mesmo positivo e negativo27.
Este estudo teve como objetivo fornecer um protocolo fácil de usar para gerar a visão de regulação transcricional (mqTrans) de um conjunto de dados do transcriptoma baseado no modelo de referência de rede grafo-atenção (GAT) pré-treinado. O framework GAT multitarefa de um trabalho publicadoanteriormente26 foi usado para transformar características transcriptômicas para características mqTrans. Um grande conjunto de dados de transcriptomas saudáveis da plataforma Xena28 da Universidade da Califórnia, Santa Cruz (UCSC) foi usado para pré-treinar o modelo de referência (HealthModel), que mediu quantitativamente as regulamentações de transcrição dos fatores regulatórios (FTs e lincRNAs) para os mRNAs alvo. A visualização mqTrans gerada pode ser usada para construir modelos de previsão e detectar biomarcadores escuros. Este protocolo utiliza o conjunto de dados de pacientes com adenocarcinoma de cólon (COAD) do banco de dados The Cancer Genome Atlas (TCGA)29 como exemplo ilustrativo. Nesse contexto, os pacientes nos estágios I ou II são categorizados como amostras negativas, enquanto aqueles nos estágios III ou IV são considerados positivos. As distribuições de biomarcadores escuros e tradicionais entre os 26 tipos de câncer TCGA também são comparadas.
Descrição do pipeline HealthModel
A metodologia empregada neste protocolo baseia-se no referencial previamentepublicado26, conforme descrito na Figura 1. Para começar, os usuários precisam preparar o conjunto de dados de entrada, alimentá-lo no pipeline HealthModel proposto e obter recursos mqTrans. Instruções detalhadas de preparação de dados são fornecidas na seção 2 da seção de protocolo. Posteriormente, os usuários têm a opção de combinar os recursos do mqTrans com os recursos transcriptômicos originais ou prosseguir apenas com os recursos do mqTrans gerados. O conjunto de dados produzido é então submetido a um processo de seleção de recursos, com os usuários tendo a flexibilidade de escolher seu valor preferido para k em validação cruzada k-fold para classificação. A principal métrica de avaliação utilizada neste protocolo é a acurácia.
O HealthModel26 categoriza as características transcriptômicas em três grupos distintos: TF (Transcription Factor), lincRNA (Long Intergenic non-coding RNA) e mRNA (RNA mensageiro). As características dos FT são definidas com base nas anotações disponíveis no Atlas de Proteínas Humanas30,31. Este trabalho utiliza as anotações de lincRNAs do conjunto de dados GTEx32. Genes pertencentes às vias de terceiro nível no banco de dados KEGG33 são considerados como características de RNAm. É importante notar que, se uma característica de RNAm exibe funções regulatórias para um gene alvo, conforme documentado no banco de dados TRRUST34, ela é reclassificada na classe TF.
Este protocolo também gera manualmente os dois arquivos de exemplo para as IDs de gene de fatores regulatórios (regulatory_geneIDs.csv) e mRNA alvo (target_geneIDs.csv). A matriz de distância par a par entre as características regulatórias (FTs e lincRNAs) é calculada pelos coeficientes de correlação de Pearson e agrupada pela popular ferramenta de análise de rede de coexpressão gênica ponderada (WGCNA)36 (adjacent_matrix.csv). Os usuários podem utilizar diretamente o pipeline HealthModel junto com esses arquivos de configuração de exemplo para gerar a exibição mqTrans de um conjunto de dados transcriptômico.
Detalhes técnicos do HealthModel
HealthModel representa as intrincadas relações entre FTs e lincRNAs como um grafo, com as características de entrada servindo como os vértices denotados por V e uma matriz de borda intervértice designada como E. Cada amostra é caracterizada por características regulatórias K , simbolizadas como VK×1. Especificamente, o conjunto de dados abrangeu 425 FTs e 375 lincRNAs, resultando em uma dimensionalidade da amostra de K = 425 + 375 = 800. Para estabelecer a matriz de borda E, este trabalho empregou a popular ferramenta WGCNA35. O peso par a par ligando dois vértices representados como 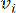 e
e 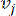 , é determinado pelo coeficiente de correlação de Pearson. A rede de regulação gênica exibe uma topologia livre de escalas36, caracterizada pela presença de genes hub com papéis funcionais fundamentais. Calculamos a correlação entre duas feições ou vértices, e , usando a medida de sobreposição topológica (TOM) da seguinte forma:
, é determinado pelo coeficiente de correlação de Pearson. A rede de regulação gênica exibe uma topologia livre de escalas36, caracterizada pela presença de genes hub com papéis funcionais fundamentais. Calculamos a correlação entre duas feições ou vértices, e , usando a medida de sobreposição topológica (TOM) da seguinte forma:
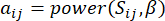 (1)
(1)
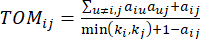 (2)
(2)
O β de limite suave é calculado usando a função ‘pickSoft Threshold’ do pacote WGCNA. A função exponencial de potência aij é aplicada, onde 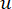 representa um gene excluindo i e j, e
representa um gene excluindo i e j, e 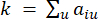 representa a conectividade de vértices. O WGCNA agrupa os perfis de expressão das características transcriptômicas em múltiplos módulos usando uma medida de dissimilaridade comumente empregada (
representa a conectividade de vértices. O WGCNA agrupa os perfis de expressão das características transcriptômicas em múltiplos módulos usando uma medida de dissimilaridade comumente empregada (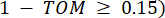 37.
37.
O framework HealthModel foi originalmente projetado como uma arquitetura de aprendizagem multitarefa26. Este protocolo utiliza apenas a tarefa de pré-treinamento do modelo para a construção da visão transcriptômica mqTrans. O usuário pode optar por refinar ainda mais o HealthModel pré-treinado sob a rede de atenção gráfica multitarefa com amostras transcriptômicas específicas de tarefas adicionais.
Detalhes técnicos da seleção e classificação de recursos
O pool de seleção de recursos implementa onze algoritmos de seleção de recursos (FS). Entre eles, três são algoritmos FS baseados em filtro: selecionando os melhores recursos K usando o Coeficiente de Informação Máxima (SK_mic), selecionando os recursos K com base no FPR do MIC (SK_fpr) e selecionando os recursos K com a maior taxa de descoberta falsa do MIC (SK_fdr). Além disso, três algoritmos FS baseados em árvore avaliam características individuais usando uma árvore de decisão com o índice de Gini (DT_gini), árvores de decisão adaptativas impulsionadas (AdaBoost) e floresta aleatória (RF_fs). O pool também incorpora dois métodos wrapper: eliminação de feições recursivas com o classificador vetorial de suporte linear (RFE_SVC) e eliminação de recursos recursivos com o classificador de regressão logística (RFE_LR). Finalmente, dois algoritmos de incorporação são incluídos: classificador linear SVC com os valores de importância de característica L1 (lSVC_L1) mais bem classificados e classificador de regressão logística com os valores de importância de característica L1 (LR_L1).
O pool de classificadores emprega sete classificadores diferentes para construir modelos de classificação. Esses classificadores compreendem máquina de vetor de suporte linear (SVC), Gaussian Naïve Bayes (GNB), classificador de regressão logística (LR), k-vizinho mais próximo, com k definido como 5 por padrão (KNN), XGBoost, floresta aleatória (RF) e árvore de decisão (DT).
A divisão aleatória do conjunto de dados no trem: subconjuntos de teste pode ser definida na linha de comando. O exemplo demonstrado usa a razão trem: teste = 8: 2.
