El transcriptoma consiste en la expresión de todos los genes en una muestra y puede ser perfilado por tecnologías de alto rendimiento como microarrays y RNA-seq1. Los niveles de expresión de un gen en un conjunto de datos se denominan característica transcriptómica, y la representación diferencial de una característica transcriptómica entre el fenotipo y los grupos control define a este gen como un biomarcador de este fenotipo 2,3. Los biomarcadores transcriptómicos se han utilizado ampliamente en las investigaciones del diagnóstico de enfermedades4, el mecanismo biológico5 y el análisis de supervivencia 6,7, etc.
Los patrones de actividad de los genes en los tejidos sanos contienen información crucial sobre las vidas 8,9. Estos patrones ofrecen información invaluable y actúan como referencias ideales para comprender las complejas trayectorias de desarrollo de los trastornos benignos10,11 y las enfermedades letales12. Los genes interactúan entre sí, y los transcriptomas representan los niveles finales de expresión después de sus complicadas interacciones. Tales patrones se formulan como la red de regulación transcripcional13 y la red metabólica14, etc. Las expresiones de ARN mensajeros (ARNm) pueden ser reguladas transcripcionalmente por factores de transcripción (TF) y ARN intergénicos largos no codificantes (lincRNAs)15,16,17. El análisis convencional de la expresión diferencial ignoró estas complejas interacciones génicas con el supuesto de independencia entre características18,19.
Los avances recientes en las redes neuronales de grafos (GNN) demuestran un potencial extraordinario en la extracción de información importante de los datos basados en OMIC para estudios de cáncer20, por ejemplo, la identificación de módulos de coexpresión21. La capacidad innata de las GNN las hace ideales para modelar las intrincadas relaciones y dependencias entre los genes22,23.
Los estudios biomédicos a menudo se centran en predecir con precisión un fenotipo en comparación con el grupo de control. Tales tareas se formulan comúnmente como clasificaciones binarias 24,25,26. Aquí, las dos etiquetas de clase se codifican normalmente como 1 y 0, verdadero y falso, o incluso positivo y negativo27.
Este estudio tuvo como objetivo proporcionar un protocolo fácil de usar para generar la vista de regulación transcripcional (mqTrans) de un conjunto de datos de transcriptoma basado en el modelo de referencia de la red de atención de grafos (GAT) preentrenado. Se utilizó el marco GAT multitarea de un trabajo publicado anteriormente26 para transformar las características transcriptómicas en las características mqTrans. Se utilizó un gran conjunto de datos de transcriptomas sanos de la plataforma Xena28 de la Universidad de California, Santa Cruz (UCSC) para preentrenar el modelo de referencia (HealthModel), que midió cuantitativamente las regulaciones de transcripción desde los factores reguladores (TF y lincRNAs) hasta los ARNm diana. La vista mqTrans generada podría utilizarse para construir modelos de predicción y detectar biomarcadores oscuros. Este protocolo utiliza el conjunto de datos de pacientes con adenocarcinoma de colon (COAD) de la base de datos del Atlas del Genoma del Cáncer (TCGA)29 como ejemplo ilustrativo. En este contexto, los pacientes en estadios I o II se clasifican como muestras negativas, mientras que los que se encuentran en estadios III o IV se consideran muestras positivas. También se comparan las distribuciones de los biomarcadores oscuros y tradicionales en los 26 tipos de cáncer TCGA.
Descripción de la canalización de HealthModel
La metodología empleada en este protocolo se basa en el marco26 publicado anteriormente, como se describe en la Figura 1. Para comenzar, los usuarios deben preparar el conjunto de datos de entrada, introducirlo en la canalización propuesta de HealthModel y obtener características de mqTrans. Las instrucciones detalladas de preparación de datos se proporcionan en la sección 2 de la sección de protocolo. Posteriormente, los usuarios tienen la opción de combinar las características de mqTrans con las características transcriptómicas originales o continuar solo con las características de mqTrans generadas. A continuación, el conjunto de datos producido se somete a un proceso de selección de características, en el que los usuarios tienen la flexibilidad de elegir su valor preferido para k en la validación cruzada de k-fold para la clasificación. La principal métrica de evaluación utilizada en este protocolo es la precisión.
HealthModel26 clasifica las características transcriptómicas en tres grupos distintos: TF (factor de transcripción), lincRNA (ARN intergénico largo no codificante) y ARNm (ARN mensajero). Las características de TF se definen en función de las anotaciones disponibles en el Atlas de Proteínas Humanas30,31. Este trabajo utiliza las anotaciones de lincRNAs del conjunto de datos GTEx32. Los genes que pertenecen a las vías de tercer nivel en la base de datos KEGG33 se consideran características de ARNm. Vale la pena señalar que si una característica de ARNm exhibe funciones reguladoras para un gen diana, como se documenta en la base de datos TRRUST34, se reclasifica en la clase TF.
Este protocolo también genera manualmente los dos archivos de ejemplo para las identificaciones de genes de los factores reguladores (regulatory_geneIDs.csv) y el ARNm diana (target_geneIDs.csv). La matriz de distancia por pares entre las características reguladoras (TFs y lincRNAs) se calcula mediante los coeficientes de correlación de Pearson y se agrupa mediante la popular herramienta de análisis de redes de coexpresión génica ponderada (WGCNA)36 (adjacent_matrix.csv). Los usuarios pueden utilizar directamente la canalización HealthModel junto con estos archivos de configuración de ejemplo para generar la vista mqTrans de un conjunto de datos transcriptómicos.
Detalles técnicos de HealthModel
HealthModel representa las intrincadas relaciones entre TF y lincRNAs como un grafo, con las características de entrada que sirven como los vértices denotados por V y una matriz de borde entre vértices designada como E. Cada muestra se caracteriza por características reguladoras K, simbolizadas como VK×1. En concreto, el conjunto de datos abarcó 425 TF y 375 lincRNAs, lo que dio como resultado una dimensionalidad de la muestra de K = 425 + 375 = 800. Para establecer la matriz de bordes E, en este trabajo se empleó la popular herramienta WGCNA35. El peso por pares que une dos vértices representados como 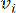 y
y 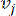 , está determinado por el coeficiente de correlación de Pearson. La red reguladora de genes exhibe una topología libre de escala36, caracterizada por la presencia de genes centrales con roles funcionales fundamentales. Calculamos la correlación entre dos características o vértices, y , utilizando la medida de superposición topológica (TOM) de la siguiente manera:
, está determinado por el coeficiente de correlación de Pearson. La red reguladora de genes exhibe una topología libre de escala36, caracterizada por la presencia de genes centrales con roles funcionales fundamentales. Calculamos la correlación entre dos características o vértices, y , utilizando la medida de superposición topológica (TOM) de la siguiente manera:
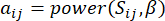 (1)
(1)
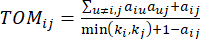 (2)
(2)
El β de umbral suave se calcula utilizando la función ‘pickSoft Threshold’ del paquete WGCNA. Se aplica la función exponencial de potencia aij , donde 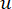 representa un gen excluyendo i y j, y
representa un gen excluyendo i y j, y 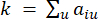 representa la conectividad de los vértices. WGCNA agrupa los perfiles de expresión de las características transcriptómicas en múltiples módulos utilizando una medida de disimilitud comúnmente empleada (
representa la conectividad de los vértices. WGCNA agrupa los perfiles de expresión de las características transcriptómicas en múltiples módulos utilizando una medida de disimilitud comúnmente empleada (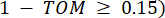 37.
37.
El marco HealthModel se diseñó originalmente como una arquitectura de aprendizaje multitarea26. Este protocolo solo utiliza la tarea de preentrenamiento del modelo para la construcción de la vista transcriptómica mqTrans. El usuario puede optar por refinar aún más el HealthModel previamente entrenado en la red de atención de grafos multitarea con muestras transcriptómicas adicionales específicas de la tarea.
Detalles técnicos de la selección y clasificación de características
El grupo de selección de características implementa once algoritmos de selección de características (FS). Entre ellos, tres son algoritmos de FS basados en filtros: selección de K mejores características utilizando el Coeficiente de Información Máxima (SK_mic), selección de K características basadas en el FPR de MIC (SK_fpr) y selección de K características con la tasa de falso descubrimiento más alta de MIC (SK_fdr). Además, tres algoritmos FS basados en árboles evalúan características individuales utilizando un árbol de decisión con el índice de Gini (DT_gini), árboles de decisión potenciados adaptativos (AdaBoost) y bosque aleatorio (RF_fs). El grupo también incorpora dos métodos contenedores: la eliminación de características recursivas con el clasificador de vectores de soporte lineal (RFE_SVC) y la eliminación de características recursivas con el clasificador de regresión logística (RFE_LR). Por último, se incluyen dos algoritmos de incrustación: el clasificador SVC lineal con los valores de importancia de la característica L1 mejor clasificados (lSVC_L1) y el clasificador de regresión logística con los valores de importancia de la característica L1 mejor clasificados (LR_L1).
El grupo de clasificadores emplea siete clasificadores diferentes para crear modelos de clasificación. Estos clasificadores comprenden la máquina de vectores de soporte lineal (SVC), Gaussian Naïve Bayes (GNB), el clasificador de regresión logística (LR), el k-vecino más cercano, con k establecido en 5 de forma predeterminada (KNN), XGBoost, bosque aleatorio (RF) y árbol de decisión (DT).
La división aleatoria del conjunto de datos en los subconjuntos train: test se puede establecer en la línea de comandos. En el ejemplo demostrado se utiliza la relación de train: test = 8:2.
