Transkriptom, bir örnekteki tüm genlerin ekspresyonlarından oluşur ve mikrodizi ve RNA-seq1 gibi yüksek verimli teknolojilerle profillenebilir. Bir veri kümesindeki bir genin ekspresyon seviyelerine transkriptomik özellik denir ve fenotip ile kontrol grupları arasındaki bir transkriptomik özelliğin diferansiyel temsili, bu geni bu fenotipin bir biyobelirteci olarak tanımlar 2,3. Transkriptomik biyobelirteçler, hastalık teşhisi4, biyolojik mekanizma5 ve sağkalım analizi 6,7 vb. araştırmalarda yaygın olarak kullanılmaktadır.
Sağlıklı dokulardaki gen aktivite paternleri yaşamlar hakkında önemli bilgiler taşır 8,9. Bu kalıplar paha biçilmez içgörüler sunar ve iyi huylu bozuklukların10,11 ve ölümcül hastalıkların12 karmaşık gelişimsel yörüngelerini anlamak için ideal referanslar olarak hareket eder. Genler birbirleriyle etkileşime girer ve transkriptomlar, karmaşık etkileşimlerinden sonraki son ekspresyon seviyelerini temsil eder. Bu tür kalıplar, transkripsiyonel düzenleme ağı13 ve metabolizma ağı14 vb. olarak formüle edilmiştir. Haberci RNA’ların (mRNA’lar) ekspresyonları, transkripsiyon faktörleri (TF’ler) ve uzun intergenik kodlamayan RNA’lar (lincRNA’lar) tarafından transkripsiyonel olarak düzenlenebilir15,16,17. Konvansiyonel diferansiyel ekspresyon analizi, özellikler arası bağımsızlık varsayımıyla bu tür karmaşık gen etkileşimlerini göz ardı etmiştir18,19.
Grafik sinir ağlarındaki (GNN’ler) son gelişmeler, kanser çalışmaları20 için OMIC tabanlı verilerden önemli bilgilerin çıkarılmasında, örneğin birlikte ekspresyon modüllerinintanımlanmasında 21 olağanüstü bir potansiyel olduğunu göstermektedir. GNN’lerin doğuştan gelen kapasitesi, onları genler arasındaki karmaşık ilişkileri ve bağımlılıkları modellemek için ideal kılar22,23.
Biyomedikal çalışmalar genellikle kontrol grubuna karşı bir fenotipi doğru bir şekilde tahmin etmeye odaklanır. Bu tür görevler genellikle ikili sınıflandırmalar 24,25,26 olarak formüle edilir. Burada, iki sınıf etiketi genellikle 1 ve 0, doğru ve yanlış, hatta pozitif ve negatif27 olarak kodlanır.
Bu çalışma, önceden eğitilmiş grafik-dikkat ağı (GAT) referans modeline dayalı bir transkriptom veri kümesinin transkripsiyonel düzenleme (mqTrans) görünümünü oluşturmak için kullanımı kolay bir protokol sağlamayı amaçlamıştır. Daha önce yayınlanmış bir çalışma26’dan çok görevli GAT çerçevesi, transkriptomik özellikleri mqTrans özelliklerine dönüştürmek için kullanıldı. Düzenleyici faktörlerden (TF’ler ve lincRNA’lar) hedef mRNA’lara transkripsiyon düzenlemelerini kantitatif olarak ölçen referans modeli (HealthModel) önceden eğitmek için Kaliforniya Üniversitesi, Santa Cruz (UCSC) Xena platform28’den sağlıklı transkriptomlardan oluşan geniş bir veri seti kullanıldı. Oluşturulan mqTrans görünümü, tahmin modelleri oluşturmak ve karanlık biyobelirteçleri tespit etmek için kullanılabilir. Bu protokol, açıklayıcı bir örnek olarak Kanser Genom Atlası (TCGA) veritabanı29’dan kolon adenokarsinomu (COAD) hasta veri setini kullanır. Bu bağlamda, evre I veya II’deki hastalar negatif örnekler olarak kategorize edilirken, evre III veya IV’teki hastalar pozitif örnekler olarak kabul edilir. 26 TCGA kanser türü arasında karanlık ve geleneksel biyobelirteçlerin dağılımları da karşılaştırılmıştır.
HealthModel işlem hattının açıklaması
Bu protokolde kullanılan metodoloji, Şekil 1’de belirtildiği gibi daha önce yayınlanmış çerçeve26’ya dayanmaktadır. Başlamak için kullanıcıların giriş veri kümesini hazırlaması, önerilen HealthModel işlem hattına beslemesi ve mqTrans özelliklerini edinmesi gerekir. Ayrıntılı veri hazırlama talimatları protokol bölümünün 2. bölümünde verilmiştir. Daha sonra, kullanıcılar mqTrans özelliklerini orijinal transkriptomik özelliklerle birleştirme veya yalnızca oluşturulan mqTrans özellikleriyle devam etme seçeneğine sahiptir. Üretilen veri kümesi daha sonra bir özellik seçim sürecine tabi tutulur ve kullanıcılar, sınıflandırma için k kat çapraz doğrulamada k için tercih ettikleri değeri seçme esnekliğine sahiptir. Bu protokolde kullanılan birincil değerlendirme ölçütü doğruluktur.
HealthModel26, transkriptomik özellikleri üç farklı gruba ayırır: TF (Transkripsiyon Faktörü), lincRNA (uzun intergenik kodlamayan RNA) ve mRNA (haberci RNA). TF özellikleri, İnsan Protein Atlası30,31’de bulunan ek açıklamalara göre tanımlanır. Bu çalışma, GTEx veri kümesi32’deki lincRNA’ların ek açıklamalarını kullanır. KEGG veri tabanındaki33 üçüncü seviye yollara ait genler, mRNA özellikleri olarak kabul edilir. Bir mRNA özelliği, TRRUST veri tabanında34 belgelendiği gibi bir hedef gen için düzenleyici roller sergiliyorsa, TF sınıfına yeniden sınıflandırıldığını belirtmekte fayda var.
Bu protokol ayrıca düzenleyici faktörlerin (regulatory_geneIDs.csv) ve hedef mRNA’nın (target_geneIDs.csv) gen kimlikleri için iki örnek dosyayı manuel olarak oluşturur. Düzenleyici özellikler (TF’ler ve lincRNA’lar) arasındaki ikili mesafe matrisi, Pearson korelasyon katsayıları ile hesaplanır ve popüler araç ağırlıklı gen ortak ekspresyon ağı analizi (WGCNA)36 (adjacent_matrix.csv) ile kümelenir. Kullanıcılar, bir transkriptomik veri kümesinin mqTrans görünümünü oluşturmak için bu örnek yapılandırma dosyalarıyla birlikte HealthModel işlem hattını doğrudan kullanabilir.
HealthModel’in teknik detayları
HealthModel, TF’ler ve lincRNA’lar arasındaki karmaşık ilişkileri bir grafik olarak temsil eder, girdi özellikleri V ile gösterilen köşeler ve E olarak gösterilen köşeler arası kenar matrisi olarak hizmet eder. Her numune, VK×1 olarak sembolize edilen K düzenleyici özelliklerle karakterize edilir. Spesifik olarak, veri kümesi 425 TF ve 375 lincRNA’yı kapsıyordu ve bu da K = 425 + 375 = 800’lük bir örnek boyutsallığı ile sonuçlandı. Kenar matrisi E’yi oluşturmak için, bu çalışma popüler araç WGCNA35’i kullandı. ve 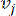 olarak
olarak 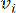 temsil edilen iki köşeyi birbirine bağlayan ikili ağırlık, Pearson korelasyon katsayısı ile belirlenir. Gen düzenleyici ağ, önemli fonksiyonel rollere sahip hub genlerinin varlığı ile karakterize edilen ölçeksiz bir topoloji36 sergiler. İki özellik veya köşe arasındaki korelasyonu ve topolojik örtüşme ölçüsünü (TOM) kullanarak aşağıdaki gibi hesaplıyoruz:
temsil edilen iki köşeyi birbirine bağlayan ikili ağırlık, Pearson korelasyon katsayısı ile belirlenir. Gen düzenleyici ağ, önemli fonksiyonel rollere sahip hub genlerinin varlığı ile karakterize edilen ölçeksiz bir topoloji36 sergiler. İki özellik veya köşe arasındaki korelasyonu ve topolojik örtüşme ölçüsünü (TOM) kullanarak aşağıdaki gibi hesaplıyoruz:
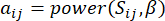 (1)
(1)
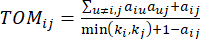 (2)
(2)
Yumuşak eşik β , WGCNA paketinden ‘pickSoft Threshold’ işlevi kullanılarak hesaplanır. Kuvvet üstel fonksiyonu aij uygulanır, burada 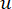 i ve j hariç bir geni temsil eder ve
i ve j hariç bir geni temsil eder ve 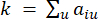 köşe bağlantısını temsil eder. WGCNA, transkriptomik özelliklerin ekspresyon profillerini, yaygın olarak kullanılan bir farklılık ölçüsü kullanarak birden fazla modülde kümeler (
köşe bağlantısını temsil eder. WGCNA, transkriptomik özelliklerin ekspresyon profillerini, yaygın olarak kullanılan bir farklılık ölçüsü kullanarak birden fazla modülde kümeler (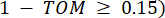 37.
37.
HealthModel çerçevesi başlangıçta çok görevli bir öğrenme mimarisiolarak tasarlanmıştır 26. Bu protokol yalnızca transkriptomik mqTrans görünümünün oluşturulması için model ön eğitim görevini kullanır. Kullanıcı, göreve özgü ek transkriptomik örneklerle çoklu görev grafiği dikkat ağı altında önceden eğitilmiş HealthModel’i daha da iyileştirmeyi seçebilir.
Özellik seçimi ve sınıflandırmasının teknik detayları
Özellik seçim havuzu, on bir özellik seçimi (FS) algoritması uygular. Bunlardan üçü filtre tabanlı FS algoritmalarıdır: Maksimum Bilgi Katsayısını (SK_mic) kullanarak K en iyi özelliklerini seçmek, MIC’nin FPR’sine (SK_fpr) göre K özelliklerini seçmek ve MIC’nin en yüksek yanlış keşif oranına sahip K özelliklerini seçmek (SK_fdr). Ek olarak, üç ağaç tabanlı FS algoritması, Gini indeksi (DT_gini), uyarlanabilir artırılmış karar ağaçları (AdaBoost) ve rastgele orman (RF_fs) içeren bir karar ağacı kullanarak bireysel özellikleri değerlendirir. Havuz ayrıca iki sarmalayıcı yöntemi içerir: Doğrusal destek vektör sınıflandırıcısı (RFE_SVC) ile özyinelemeli özellik eleme ve lojistik regresyon sınıflandırıcısı (RFE_LR) ile özyinelemeli özellik eleme. Son olarak, iki ekleme algoritması dahil edilmiştir: en üst sıradaki L1 özellik önem değerlerine (lSVC_L1) sahip doğrusal SVC sınıflandırıcısı ve en üst sıradaki L1 özellik önem değerlerine (LR_L1) sahip lojistik regresyon sınıflandırıcısı.
Sınıflandırıcı havuzu, sınıflandırma modelleri oluşturmak için yedi farklı sınıflandırıcı kullanır. Bu sınıflandırıcılar, doğrusal destek vektör makinesi (SVC), Gauss Naïve Bayes (GNB), lojistik regresyon sınıflandırıcısı (LR), k-en yakın komşu, k varsayılan olarak 5’e ayarlanmış (KNN), XGBoost, rastgele orman (RF) ve karar ağacından (DT) oluşur.
Veri kümesinin trene rastgele bölünmesi: test alt kümeleri komut satırında ayarlanabilir. Gösterilen örnek, tren: test = 8: 2 oranını kullanır.
