Le transcriptome est constitué de l’expression de tous les gènes d’un échantillon et peut être profilé par des technologies à haut débit telles que les puces à ADN et le séquençage de l’ARN1. Les niveaux d’expression d’un gène dans un ensemble de données sont appelés une caractéristique transcriptomique, et la représentation différentielle d’une caractéristique transcriptomique entre le phénotype et le groupe témoin définit ce gène comme un biomarqueur de ce phénotype 2,3. Les biomarqueurs transcriptomiques ont été largement utilisés dans les enquêtes sur le diagnostic de la maladie4, le mécanisme biologique5 et l’analyse de survie6, 7, etc.
Les modèles d’activité des gènes dans les tissus sains transportent des informations cruciales sur la vie 8,9. Ces modèles offrent des informations précieuses et constituent des références idéales pour comprendre les trajectoires développementales complexes des troubles bénins10,11 et des maladies mortelles12. Les gènes interagissent les uns avec les autres, et les transcriptomes représentent les niveaux d’expression finaux après leurs interactions compliquées. De tels modèles sont formulés comme le réseau de régulation transcriptionnelle13 et le réseau métabolique14, etc. L’expression des ARN messagers (ARNm) peut être régulée transcriptionnellement par des facteurs de transcription (TF) et de longs ARN intergéniques non codants (ARNlinc)15,16,17. L’analyse conventionnelle de l’expression différentielle a ignoré ces interactions complexes entre les gènes avec l’hypothèse de l’indépendance inter-caractéristiques18,19.
Les progrès récents dans les réseaux neuronaux à graphes (GNN) démontrent un potentiel extraordinaire dans l’extraction d’informations importantes à partir de données OMIC pour les études sur le cancer20, par exemple, l’identification de modules de co-expression21. La capacité innée des GNN les rend idéaux pour modéliser les relations et les dépendances complexes entre les gènes22,23.
Les études biomédicales se concentrent souvent sur la prédiction précise d’un phénotype par rapport au groupe témoin. De telles tâches sont généralement formulées sous forme de classifications binaires 24,25,26. Ici, les deux étiquettes de classe sont généralement codées comme 1 et 0, vrai et faux, ou même positif et négatif27.
Cette étude visait à fournir un protocole facile à utiliser pour générer la vue de régulation transcriptionnelle (mqTrans) d’un ensemble de données de transcriptomes basé sur le modèle de référence du réseau d’attention graphique pré-entraîné (GAT). Le cadre GAT multitâche d’un travail précédemment publié26 a été utilisé pour transformer les caractéristiques transcriptomiques en caractéristiques mqTrans. Un grand ensemble de données de transcriptomes sains provenant de la plate-forme Xena28 de l’Université de Californie à Santa Cruz (UCSC) a été utilisé pour pré-entraîner le modèle de référence (HealthModel), qui a mesuré quantitativement les régulations de transcription des facteurs régulateurs (TF et ARNlinc) aux ARNm cibles. La vue mqTrans générée pourrait être utilisée pour construire des modèles de prédiction et détecter des biomarqueurs sombres. Ce protocole utilise l’ensemble de données de patients atteints d’adénocarcinome du côlon (COAD) de la base de données29 de l’Atlas du génome du cancer (TCGA) à titre d’exemple. Dans ce contexte, les patients des stades I ou II sont classés comme des échantillons négatifs, tandis que ceux des stades III ou IV sont considérés comme des échantillons positifs. Les distributions des biomarqueurs sombres et traditionnels dans les 26 types de cancer TCGA sont également comparées.
Description du pipeline HealthModel
La méthodologie employée dans ce protocole est basée sur le cadre26 publié précédemment, comme indiqué à la figure 1. Pour commencer, les utilisateurs doivent préparer le jeu de données d’entrée, l’introduire dans le pipeline HealthModel proposé et obtenir des fonctionnalités mqTrans. Des instructions détaillées pour la préparation des données sont fournies à la section 2 de la section sur le protocole. Par la suite, les utilisateurs ont la possibilité de combiner les caractéristiques mqTrans avec les caractéristiques transcriptomiques d’origine ou de procéder uniquement avec les caractéristiques mqTrans générées. Le jeu de données produit est ensuite soumis à un processus de sélection de caractéristiques, les utilisateurs ayant la possibilité de choisir leur valeur préférée pour k dans la validation croisée k fois pour la classification. La principale mesure d’évaluation utilisée dans ce protocole est la précision.
HealthModel26 classe les caractéristiques transcriptomiques en trois groupes distincts : TF (facteur de transcription), lincRNA (long ARN intergénique non codant) et ARNm (ARN messager). Les caractéristiques TF sont définies sur la base des annotations disponibles dans l’Atlas des protéines humaines30,31. Ce travail utilise les annotations des ARNlinc du jeu de données GTEx32. Les gènes appartenant aux voies de troisième niveau de la base de données KEGG33 sont considérés comme des caractéristiques de l’ARNm. Il convient de noter que si une caractéristique de l’ARNm présente des rôles régulateurs pour un gène cible, comme documenté dans la base de données TRRUST34, elle est reclassée dans la classe TF.
Ce protocole génère également manuellement les deux fichiers d’exemple pour les identifiants de gènes des facteurs de régulation (regulatory_geneIDs.csv) et de l’ARNm cible (target_geneIDs.csv). La matrice de distance par paires entre les caractéristiques régulatrices (TF et ARNlinc) est calculée par les coefficients de corrélation de Pearson et regroupée par l’outil populaire WGCNA (Weighted Geme Co-Expression Network Analysis)36 (adjacent_matrix.csv). Les utilisateurs peuvent utiliser directement le pipeline HealthModel avec ces exemples de fichiers de configuration pour générer la vue mqTrans d’un jeu de données transcriptomique.
Détails techniques de HealthModel
HealthModel représente les relations complexes entre les TF et les lincRNAs sous la forme d’un graphique, les entités en entrée servant de sommets notés V et une matrice d’arêtes inter-sommets désignée par E. Chaque échantillon est caractérisé par des caractéristiques régulatrices K , symbolisées par VK×1. Plus précisément, l’ensemble de données comprenait 425 TF et 375 ARNlinc, ce qui donne une dimensionnalité de l’échantillon de K = 425 + 375 = 800. Pour établir la matrice d’arête E, ce travail a utilisé l’outil populaire WGCNA35. Le poids par paire reliant deux sommets représentés par 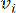 et
et 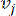 , est déterminé par le coefficient de corrélation de Pearson. Le réseau de régulation des gènes présente une topologie sans échelle36, caractérisée par la présence de gènes pivots ayant des rôles fonctionnels pivots. Nous calculons la corrélation entre deux entités ou sommets, et , à l’aide de la mesure de chevauchement topologique (TOM) comme suit :
, est déterminé par le coefficient de corrélation de Pearson. Le réseau de régulation des gènes présente une topologie sans échelle36, caractérisée par la présence de gènes pivots ayant des rôles fonctionnels pivots. Nous calculons la corrélation entre deux entités ou sommets, et , à l’aide de la mesure de chevauchement topologique (TOM) comme suit :
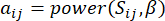 (1)
(1)
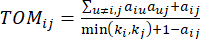 (2)
(2)
Le β de seuil souple est calculé à l’aide de la fonction « pickSoft Threshold » du package WGCNA. La fonction exponentielle de puissance aij est appliquée, où 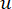 représente un gène excluant i et j, et
représente un gène excluant i et j, et 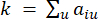 représente la connectivité du sommet. WGCNA regroupe les profils d’expression des caractéristiques transcriptomiques en plusieurs modules à l’aide d’une mesure de dissimilarité couramment utilisée (
représente la connectivité du sommet. WGCNA regroupe les profils d’expression des caractéristiques transcriptomiques en plusieurs modules à l’aide d’une mesure de dissimilarité couramment utilisée (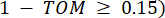 37.
37.
Le cadre HealthModel a été conçu à l’origine comme une architecture d’apprentissage multitâche26. Ce protocole utilise uniquement la tâche de pré-entraînement du modèle pour la construction de la vue transcriptomique mqTrans. L’utilisateur peut choisir d’affiner davantage le HealthModel pré-entraîné dans le cadre du réseau d’attention graphique multitâche avec des échantillons transcriptomiques supplémentaires spécifiques à la tâche.
Détails techniques de la sélection et de la classification des caractéristiques
Le pool de sélection de caractéristiques implémente onze algorithmes de sélection de caractéristiques (FS). Parmi eux, trois sont des algorithmes FS basés sur des filtres : la sélection de K meilleures caractéristiques à l’aide du coefficient d’information maximal (SK_mic), la sélection de K caractéristiques en fonction du FPR de MIC (SK_fpr) et la sélection de K entités avec le taux de fausses découvertes le plus élevé de MIC (SK_fdr). De plus, trois algorithmes FS basés sur des arbres évaluent les caractéristiques individuelles à l’aide d’un arbre de décision avec l’indice de Gini (DT_gini), d’arbres de décision adaptatifs boostés (AdaBoost) et d’une forêt aléatoire (RF_fs). Le pool intègre également deux méthodes d’encapsulation : l’élimination de caractéristiques récursives avec le classificateur de vecteurs de support linéaire (RFE_SVC) et l’élimination de caractéristiques récursives avec le classificateur de régression logistique (RFE_LR). Enfin, deux algorithmes d’incorporation sont inclus : le classificateur SVC linéaire avec les valeurs d’importance des caractéristiques L1 les mieux classées (lSVC_L1) et le classificateur de régression logistique avec les valeurs d’importance des caractéristiques L1 les mieux classées (LR_L1).
Le pool de classificateurs utilise sept classificateurs différents pour créer des modèles de classification. Ces classificateurs comprennent la machine à vecteurs de support linéaire (SVC), le bayésien naïf gaussien (GNB), le classificateur de régression logistique (LR), le k plus proche voisin, avec k défini sur 5 par défaut (KNN), XGBoost, la forêt aléatoire (RF) et l’arbre de décision (DT).
La division aléatoire du jeu de données en sous-ensembles train : test peut être définie dans la ligne de commande. L’exemple illustré utilise le rapport de train : test = 8 : 2.
