The present study validated the cytosolic biosensor in both plate reader and microscope assays. Once cells expressed the biosensor, they were stimulated with either 10 µM forskolin (a direct activator of adenylyl cyclase), 10 nM isoproterenol (an agonist at ß1AR and ß2AR), or vehicle (Figure 1). The subsequent changes in fluorescence, indicative of cAMP production, were captured every 30 s.
The data was transformed as the change in fluorescence from the initial fluorescence (ΔF/F0). A one-site decay model was applied to the fluorescence data to quantify the temporal changes in cAMP levels across the different conditions (Figure 2 and Figure 3). The parameters of these decay curves can be used to quantify the response to a given concentration of the drug. The product of the decay rate (k) and the plateau as a single value that quantifies the drug response was used, and these were used to create concentration-response curves when multiple concentrations of the same drug are applied13.
On the plate reader, upon stimulation with 10 µM forskolin, the cytosolic sensor showed a sizable decrease in fluorescence intensity within seconds and progressed over many minutes (from the baseline 0 ΔF/Fo at 200 s to -0.7 ΔF/Fo at 600 s) in HEK-293 cells (Figure 2A). Given the green sensor's design, a decrease in fluorescence indicates an increase in cAMP production, indicating that forskolin caused a uniform increase in cAMP levels in the cytosol. Stimulating HEK-293 cells with a sub-maximal concentration of isoproterenol (10 nM) led to rapid but smaller decreases in the biosensor fluorescence (-0.3 ΔF/Fo at 600 s, Figure 2A). When these lower concentrations of isoproterenol (10 nM and 100 nM) were examined in a single well of cells over time, oscillations of cAMP levels were observed in HEK-293 cells (Figure 2B). The periodicity of these oscillations was consistently around 200 s. The routine data analysis includes fitting the response to a single site decay model, which averages these oscillations (see connecting line). Averaging multiple experiments together typically led to these oscillations becoming obscured in the data. Nonetheless, the biosensor displays a sensitivity and rapid kinetics that allow the observation of cAMP oscillations.
cADDis can also be measured using a fluorescence microscope. This approach allows monitoring of cAMP in single cells and can also be adapted to visualize biosensors that are targeted to different subcellular locations. In the present study, the biosensor was used in primary human airway smooth muscle (HASM) cells. Stimulation of HASM with either vehicle, 10 µM forskolin, or 10 nM isoproterenol leads to an observable decrease in fluorescence intensity over time (from the baseline 0 ΔF/Fo at 120 s to -0.9 ΔF/Fo at 410 s) (Figure 3) (Video 1, Video 2, and Video 3). Fluorescence is evenly distributed throughout the cytosol of cells and excludes the nucleus (see the time-lapse videos). Thus, forskolin and isoproterenol produced a similar rapid and sizable decrease in fluorescence HASM cells. These data demonstrate the ability to use the biosensor in primary cell cultures.
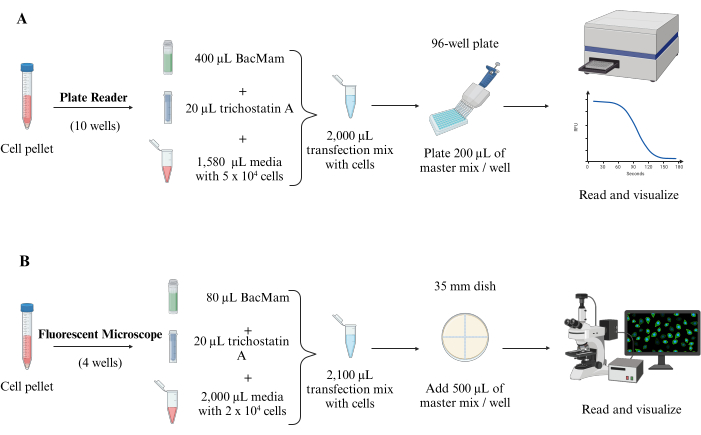
Figure 1: Schematic representation of the biosensor protocol steps. Initially, culture cells are split and resuspended with fresh media, then infected with the BacMam virus carrying the cADDis sensors. Post-transfection cells are stimulated with pharmacological agents triggering cellular pathways that alter cAMP. Changes in the fluorescence intensity in response to the varying cAMP concentration are captured using a plate reading spectrophotometer (A) or fluorescence microscope (B), providing a real-time quantification of the cAMP dynamics. Please click here to view a larger version of this figure.
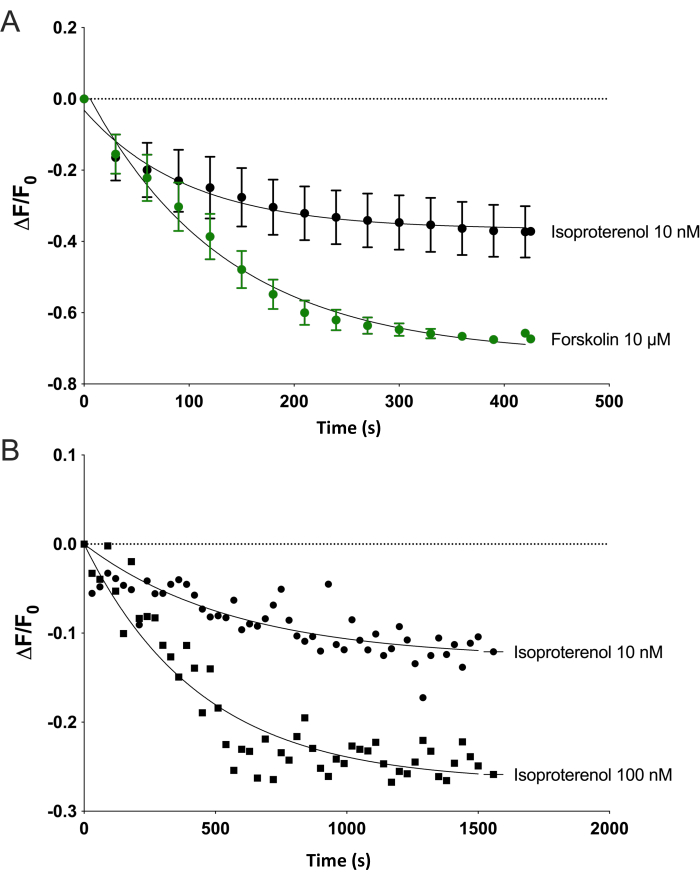
Figure 2: Real-time monitoring of cAMP in live HEK-293 cells on a plate reading spectrophotometer. Fluorescence of HEK-293 cells on 96-well plates expressing the biosensor was measured over time. After baseline fluorescence was established, fluorescence decay was monitored for 7 min. (A) Fluorescence decay after the addition of either 10 µM forskolin or 10 nM of isoproterenol is plotted as the mean ± SEM of 10-15 experiments. (B) Fluorescence decay oscillations are shown by plotting a single experiment after the addition of either 10 nM or 100 nM of isoproterenol. Data in both graphs is fit to a single site decay model (connecting line). Please click here to view a larger version of this figure.
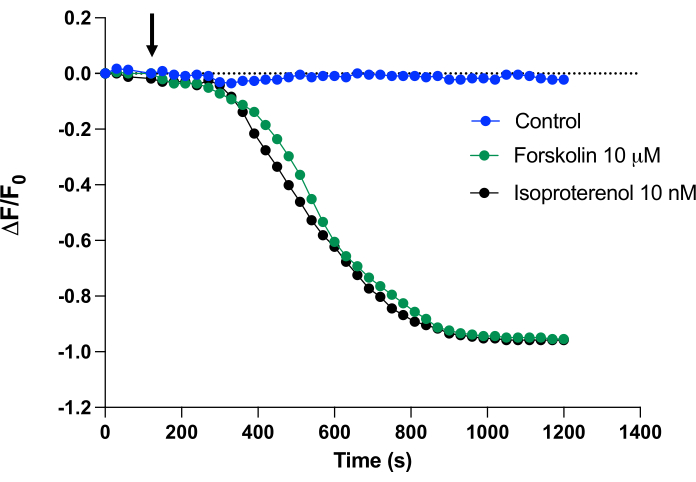
Figure 3: Real-time monitoring of cAMP in live HASM cells on a fluorescence microscope. Fluorescence of HASM cells on plated 35 mm dishes expressing the biosensor was measured over time. After baseline values were established, fluorescence decay was measured every 30 s for 20 min. The indicated agent was added at 120 s (arrow). The biosensor fluorescence decay curves in response to either vehicle, 10 µM forskolin, or 10 nM isoproterenol. Data from a single experiment is shown with a connecting line. Time-lapse videos of cells treated with vehicle (control), forskolin, or isoproterenol are included to provide a visual representation of the fluorescent responses of the biosensor. Please click here to view a larger version of this figure.
Video 1: Time-lapse video of real-time monitoring of cAMP in live HASM cells in response to the vehicle. Fluorescence decay curves of the biosensor in response to the vehicle (control). Each frame was captured every 30 s for 20 min. Scale bar: 40 μM. Please click here to download this Video.
Video 2: Time-lapse video of real-time monitoring of cAMP in live HASM cells in response to 10 µM forskolin. Fluorescence decay curves of the biosensor in response to 10 µM forskolin. Each frame was captured every 30 s for 20 min. Scale bar: 40 μM. Please click here to download this Video.
Video 3: Time-lapse video of real-time monitoring of cAMP in live HASM cells in response to 10 nM isoproterenol. Fluorescence decay curves of the biosensor in response to 10 nM isoproterenol. Each frame was captured every 30 s for 20 min. Scale bar: 40 μM. Please click here to download this Video.
| Media formulation | |
| HASM medium | |
| Name | Volume |
| Ham's F-12K | 419.65 mL |
| FBS | 50 mL |
| HEPES (1M) | 12.5 mL |
| Sodium hydroxide solution | 6 mL |
| L-glutamine 200 mM (100X) | 5 mL |
| Calcium chloride (1 M) | 0.850 mL |
| Antibiotic-Antimycotic (100X) | 5 mL |
| Primocin | 1 mL |
| HEK medium | |
| Name | Volume |
| DMEM (1x) | 444 mL |
| FBS | 50 mL |
| Antibiotic-Antimycotic (100X) | 5 mL |
| Primocin | 1 mL |
Table 1: Media formulation of HASM medium and HEK medium.
