1. Preparations for yeast transformation
This protocol is designed for ten 96-well plates but can be scaled up or down accordingly. We have found that this protocol does not work well for more than twenty 96-well plates per round of transformation. The entire transformation procedure (from step I.3) will take approximately eight hours.
- Aliquot 5-10μL of plasmid DNA (100 ng/μl) from the Yeast FLEXGene ORF library into each well of a round-bottom 96-well plate with Biorobot RapidPlate liquid handler. Place uncovered 96-well plates in clean bench fume hood overnight to dry. We have found similar transformation efficiencies with CEN and 2μ yeast plasmids.
- Inoculate 200mL of yeast peptone dextrose (YPD) media (10g/L yeast extract, 20g/L peptone, 20g/L glucose) with query strain in a 500mL baffled Erlenmeyer flask. Incubate overnight at 30°C with shaking (200 rpm). If required, selective media can also be used for these starter cultures.
- The following morning, dilute the query strain to OD600=0.1 in 2L of YPD. Shake for 4-6 hours at 30°C (200 rpm) until the culture reaches OD600=0.8-1.0. For optimal growth, grow up two 1L cultures in separate 2.8 L baffled flasks. If required, selective media can also be used instead of YPD.
2. Yeast transformation
- Harvest cells by centrifugation. Fill four disposable 225mL conical tubes with yeast culture and spin at 3000rpm (˜1800 x g) for 5 minutes in tabletop centrifuge (Eppendorf 5810R). Pour off supernatant and repeat until all cells are harvested.
- Wash cells in 200mL of autoclaved distilled H2O. Resuspend cell pellet by shaking or vortexing. Combine cells in one 225mL conical tube. Spin at 3000rpm for 5 minutes. Remove supernatant.
- Prepare 0.1MLiOAc/1XTE solution (0.1M LiOAc, 10mM Tris, 1mM EDTA, pH 8 in autoclaved distilled water). Wash cells in 100mL 0.1MLiOAc/1XTE. Spin at 3000rpm for 5 minutes. Remove supernatant.
- Resuspend cell pellet in 70mL 0.1M LiOAc/1xTE. Shake and/or vortex to ensure pellet is completely resuspended.
- Incubate with shaking at 30°C for 30 minutes (200 rpm).
- Add β-mercaptoethanol to 0.1M. Incubate with shaking at 30°C for 30 minutes (200 rpm). Note: This step can be omitted if using yeast strains that are sensitive to β-mercaptoethanol. We have found that this step is not essential for successful transformation, however it does improve transformation efficiency.
- Boil 2mL of sonicated salmon sperm DNA (10mg/mL) for 15 minutes, and then chill on ice.
- Add 2mL of boiled salmon sperm DNA to cell mixture. Caution: A precipitate may form upon addition of the salmon sperm DNA to the cell mixture. Using a pipetman and 200 μl tip, carefully remove any precipitate that forms because this may interfere with downstream pipetting.
- Aliquot 50μL of cell mixture to each well of the 96-well plate using the BioRobot Rapidplate (can also use multi-channel pipette). Do not mix.
- Incubate at room temperature for 30 minutes without shaking.
- Prepare 200mL of 40%PEG-3350/10%DMSO/0.1M LiOAc (160mL of 50% PEG-3350, 20mL DMSO, 20mL 1M LiOAc). Prepare solution immediately before use.
- Add 125μL of PEG/LioAc/DMSO solution to each well. Mix by aspirating and dispensing cell suspension eight times with RapidPlate.
- Incubate at room temperature for 30 minutes without shaking.
- Heat shock the cells at 42°C for 15 minutes in a dry incubator. Do not stack plates. Note: We have found that this heat shock step is not essential for successful transformation. For certain yeast strains (for example, temperature sensitive mutants), heat shock is deleterious. We have previously reduced the heat shock time to one minute and have been able to successfully transform a yeast strain harboring a ypt1 temperature sensitive mutation using this protocol.
- Spin plates at 2500rpm (˜1100 x g) for 5 minutes, using centrifuge adapters to accommodate 96-well plates. Remove PEG solution by inverting plates over waste bucket. Blot inverted plate on paper towel to remove residual liquid (cells will remain on bottom of wells).
- Rinse cells by adding 200μL of minimal media (e.g. SD/-Ura) to each well with RapidPlate.
- Spin plates at 2500rpm for 5 minutes and remove supernatant by inverting over waste bucket and blotting on paper towels.
- Add 200μL of minimal media (e.g. SD/-Ura) to each well with RapidPlate.
- Incubate at 30°C for 2 days without shaking. After 2 days, small colonies of cells should begin to form at the bottom of each successfully transformed well.
3. Spotting assay
- Dispense 200μL of raffinose based minimal media (e.g. SRaf/-Ura) into each well of a new flat-bottom 96-well plate.
- Mix each well of transformation plate by aspirating and dispensing cell suspension eight times with Rapidplate.
- Inoculate SRaf plates with 5μL of cell mixture from transformation plate.
- Incubate at 30°C for one day. Small colonies should be present at the bottom of each well after one day.
- Spot colonies on SD/-Ura and SGal/-Ura plates in hood. Sterilize 96-bolt replicator (frogger) by flaming. Mix colonies with frogger before spotting on selective media plates. After spotting, allow plates to dry in hood for 5-10 minutes.
- Incubate at 30°C for 2-3 days.
Photograph plates with digital camera, and visually compare growth of colonies on SGal/-Ura plates to find colonies in which query strain toxicity is enhanced (slower growth/less dense colonies) or suppressed (faster growth/more dense colonies).
4. Representative results:
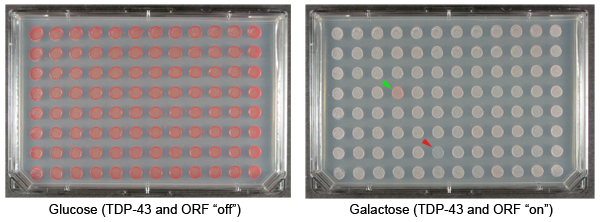
Figure 1. Yeast plasmid overexpression screen to identify suppressors and enhancers of TDP-43 toxicity. TDP-43 is a human protein that has been implicated in the pathogenesis of amyotrophic lateral sclerosis (Lou Gehrig’s disease). Cytoplasmic aggregates of TDP-43 accumulate in the brain and spinal cord neurons of ALS patients 17. Expressing TDP-43 in yeast cells results in aggregation and cytotoxicity 18. We have used this model system to define mechanisms of TDP-43 toxicity 19,20. Shown are repesentative examples of plates from our yeast TDP-43 toxicity modifier screen. These plates display colonies with an integrated galactose-inducible TDP-43 plasmid and also transformed with plasmids from the FLEXGene ORF expression library. The plate on the left contains glucose, which represses expression of TDP-43 or the FLEXGene plasmids. The plate on the right contains galactose, which induces the expression of TDP-43 and the ORFs in the FLEXGene plasmids. The green arrowhead indicates a colony transformed with a plasmid that suppresses the toxicity of TDP-43. The red arrowhead indicates a colony transformed with a plasmid that enhances the toxicity of TDP-43.