Long-term time-lapse microscopy and direct longitudinal tracking allows for the study of many anti-cancer effects during drug response. Following the general outline in Figure 1, multiple examples of cells are shown expressing validated fluorescent reporters that treated with anti-cancer drugs, tracked, and analyzed using different approaches.
Phase contrast microscopy alone is very informative and robustly reports on interphase versus mitosis, mitotic duration and arrest, abnormal cell division, and cell death21,23,24. Drugs that target cell division, often termed anti-mitotic drugs, continue to be developed. Figure 2 and Movies 1 and 2 show examples of a pair of matched breast cancer-derived MCF7 cell lines that differ only in their p53 status, treated with 500 nM of a type of anti-cancer drug that targets and inhibits the mitotic motor protein, Kinesin-5 (KSP1, Kif11, Eg5), resulting in protracted mitosis20. Wild type MCF7 cells (Figure 2A) are a paradigm to study p53-dependent cell cycle arrest25-27. Wild type cells enter mitosis, remain for several hours, eventually leave and largely arrest with induction of p5325. When p53 is removed by stable p53 knockdown (MCF7 sh p53), instead of arresting after they leave mitosis, the cells go through repeated cycles (Figure 2B). Cells were tracked manually and the mitotic index and the percentage of cells that enter a second mitosis were scored (Figure 2C, D). We observe that the sh p53 cell tracked divides when it enters a second round of mitosis rather than arresting and leaving mitosis without division. While not shown here the duration of mitotic events, time between successive mitoses, percent of cell divisions, and associated cell death events can also be scored20,21.
The taxanes, for example paclitaxel and docetaxel, are common chemotherapy for many cancers, including those that are difficult to treat like pancreatic and advanced breast. Paclitaxel binds to the dynamic plus-end of microtubules and stabilizes them, preventing their normal function. Paclitaxel has noted dose-dependent effects, and even at low concentrations can perturb normal mitotic progression and chromosome segregation16. Faithful segregation of chromosomes is essential to normal cell proliferation and when abnormal can result in aneuploidy that may trigger cell cycle arrest, but also act as a driver of cancer progression. Figure 3 and Movie 3 show cervical carcinoma-derived HeLa cells stably expressing the chromatin marker histone-2b fused to mCherry and beta-tubulin fused to EGFP (not shown) in normal growth medium treated with 1 nM paclitaxel. In this example, entry into and progression through mitosis can be followed. The timing of mitosis appears normal in this cell, however chromosome alignment and segregation is not, resulting in nuclear bulges and micronuclei that are structures indicating poor chromosome segregation. Micronuclei are prone to DNA damage and chromothripsis, which is the large-scale fragmentation of chromosomes or chromatin – this has important implications in tumor evolution28,29. While not shown here, the origin of micronuclei with relation to other mitotic structures and the fate of these cells can be directly tracked using long-term time-lapse. Further, a chromatin marker expressed with a DNA damage reporter could be used to establish the relationship between chromosome segregation, micronuclei and DNA damage.
Fluorescent reporters allow for enumerable cell processes to be tracked and new reporters are continually being developed. For example, the cell cycle consists of phases that are of particular interest in developing targeted anti-cancer therapeutics. Figure 4 and Movie 4 shows a fibrosarcoma-derived cell line (HT1080) that stably co-expresses two reporters termed, fluorescent ubiquitin cell cycle indicators (FUCCI)30. In the system here, a portion of the Cdt1 polypeptide is fused to mKO2 (monomeric Kusabira orange 2) and increases in G1-phase and is degraded in early S-phase, and a portion of geminin is fused to mAG (monomeric Azami green) and increases in mid-S-phase and is degraded upon anaphase. This cell is in normal growth medium and progresses through the cell cycle in 15 hr. Figure 5A, B and Movie 5 show the same cells in normal medium treated with 10 µM PD0332991, a Cdk4/6 inhibitor. The cells progress through G2-phase and divide normally, and strongly arrest in the subsequent G1-phase, indicating the potential for effective cytostatic effects in growing tumors. Figure 5C, D and Movie 6 show the same cells in normal medium treated with a small molecule called selinexor (KPT-330), a potent inhibitor the nuclear export protein, exportin-1 (XPO1, a.k.a. CRM1). These compounds are termed selective inhibitors of nuclear export (SINE) and their anti-cancer effects are currently under investigation31,32. SINE treatment results in strong cell cycle phenotypes and cell death33,34. This example shows a cell that progresses through G1-phase with normal kinetics (approximately 6 hr), but experiences delay in S-phase progression as indicated by the period with both red and green signal (approximately 3 hr in control but 10 in SINE treated). This cell dies in late S- or G2-phase after 21 hr 30 min; a normal cell cycle is approximately 15 hr. The effects of selinexor are being studied for different blood and solid tumors35.
A mainstay of anti-cancer therapy is cytotoxicity through catastrophic DNA damage. DNA damage can be induced through many therapies including radiation, platinum-based adducts, and small molecule drugs – for example, those that target topoisomerase I and/or II. Many combination therapies also attack the DNA damage axis by either inducing damage through separate pathways or blocking the ability of cells to repair the damage. The kinetics and level of damage and if and how this causes cell death is of widespread importance in developmental therapeutics. Figure 6 and Movie 7 show HT1080 cells that stably express a double-strand DNA damage reporter, mCherry-BP1-236 in normal growth medium treated with 10 µM etoposide (VP-16), a topoisomerase II poison. This reporter is comprised of a portion of the DNA double-strand break site protein, 53BP1 that is fused to mCherry. The nucleus of this cell was tracked using the Analyze Particles plugin in ImageJ and the integrated mCherry-BP1-2 signal was measured in each frame after thresholding out values that eliminated soluble nuclear probe. DNA damage is minimal for the first 10 hr and then steadily increases. Topoisomerase II inhibitors are known to particularly affect S- and G2-phase, when the enzyme is most active37,38. The kinetics observed in this example could indicate cell cycle associated damage; combining mCherry-BP1-2 with FUCCI reporters could demonstrate the timing of the damage that can then be linked to cell fate.
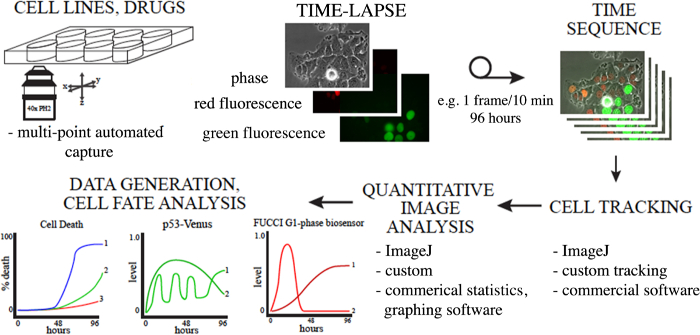
Figure 1. Overview of Using Long-term Time-lapse Microscopy and Longitudinal Tracking to Study Anti-cancer Drug Response. Cells in appropriate live-cell imaging dishes labeled as desired are imaged, cells or regions of interest are tracked, and the data is analyzed. Many methods exist to track and quantify cells, some are indicated here. Please click here to view a larger version of this figure.
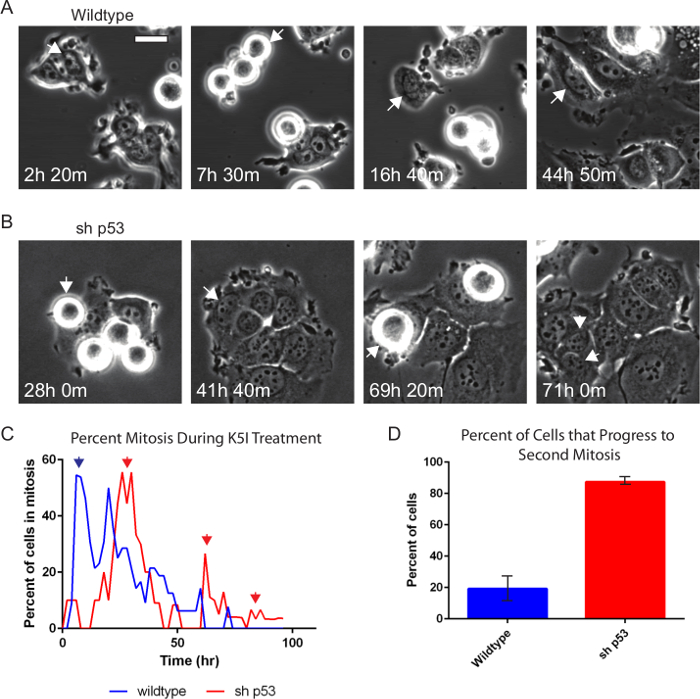
Figure 2. Phase-contrast Time-lapse Microscopy Shows p53-dependence after Anti-mitotic Drug Treatment. Wildtype (A) and p53-knockdown (B) MCF7 cells were treated with 500 nM Kinesin-5 inhibitor and imaged every 10 min for 96 hr using phase-contrast microscopy with a 20X PH2 0.70 NA lens. Individual cells were tracked manually and the percent mitosis and if the cell progresses to another mitosis again during the time-lapse were scored. Arrows indicate cell that is being tracked. The sh p53 cell (B) divides upon entering a second mitosis. (C) Both cell lines show prolonged mitotic arrest as indicated by the high peak percent mitosis (first blue and red arrows). (C, D) Nearly 90% of cells without p53 (sh p53, n = 87) show continued progression (red arrows) compared to 20% of wildtype (n = 130). Error bars indicate standard deviation. Bar = 20 µm. Movies 1 and 2. Please click here to view a larger version of this figure.
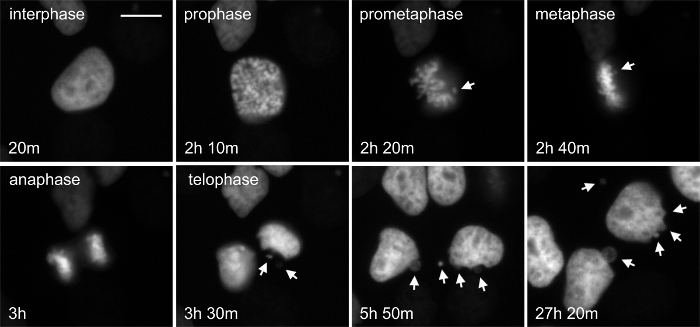
Figure 3. The Chromatin Marker Histone 2b Reveals Evidence of Chromosome Segregation Abnormalities after Low-dose Paclitaxel Treatment. Paclitaxel is a microtubule-targeted drug that results in complex, concentration-dependent defects in cell growth and division. The organization of the chromatin informs on different cell states, including the stage of mitosis and cell death. HeLa cells stably expressing both H2b-mCherry and β-tubulin-EGFP were treated with 1 nM paclitaxel. This cell is initially in interphase, progresses through the stages of mitosis and divides. While the time in mitosis appears normal, there is evidence of chromosome attachment and segregation errors that are resolved (arrows). The fate of these cells can be determined directly by longitudinal tracking. Phase-contrast (not shown) and fluorescent images were acquired at 1 frame per 10 min with a 20X Ph2 0.70 NA lens. Bar = 10 µm. Movie 3. Please click here to view a larger version of this figure.
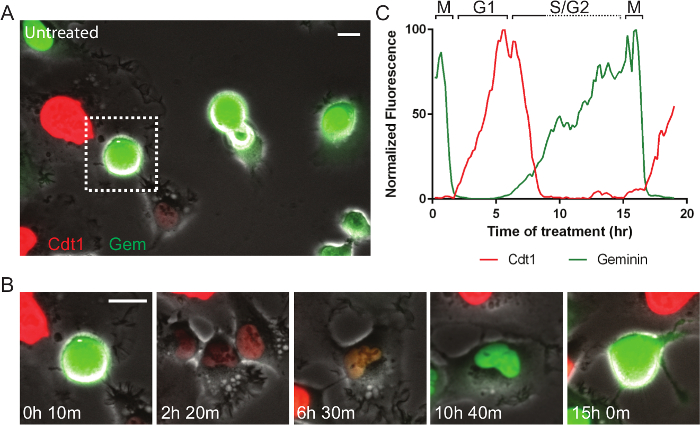
Figure 4. Fluorescent Cell Cycle Markers Allow for Direct Monitoring of Cell Cycle Progression. (A) A field of live HT1080 fibrosarcoma cells expressing the FUCCI system imaged by phase-contrast and fluorescence. (B, C) The mitotic cell in the dashed box in panel A is followed. Normally progress through the cell cycle in approximately 15 hr. After mitosis, cells are briefly dim, and then become red as they progress into and through G1-phase. When cells enter S-phase, the red Cdt1 probe is degraded and the green geminin probe increases. The brief approximately 3 hr period where both probes are present, indicates early S-phase. As cells progress through S- and G2-phase and into the next mitosis they remain green. The green probe is degraded upon anaphase of cell division. Phase-contrast and fluorescent images were acquired at 1 frame per 10 min with a 20X PH2 0.70 NA lens. Bar = 10 µm. Movie 4. Please click here to view a larger version of this figure.
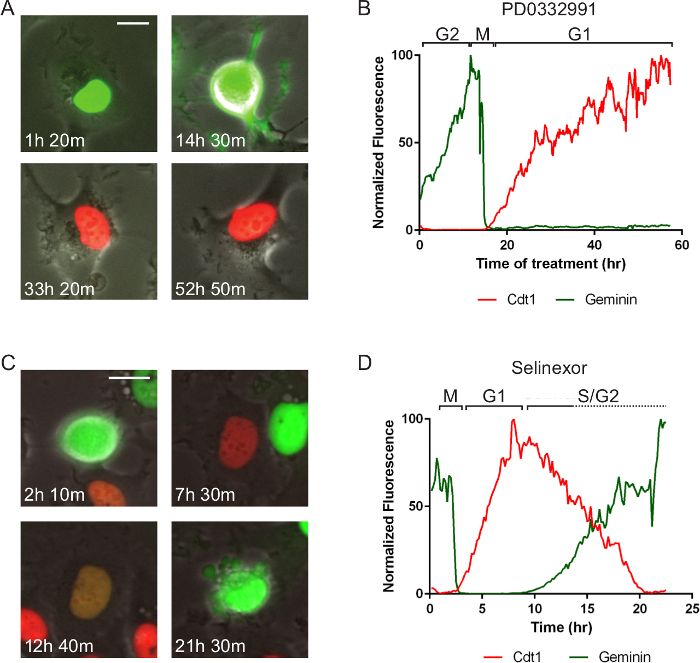
Figure 5. Cell Cycle Specific Effects and Associated Cell Death. The same cell line as in Figure 4 but treated with two different molecules that represent different anti-cancer targets. Times after treatment are indicated. (A, B) After treatment of a late S/G2-phase cell with 10 µM PD0332991 Cdk4/6 inhibitor, it cell progresses normally to mitosis (M) and divides. One daughter cell is tracked by measuring red and green fluorescent intensities in the region of interest in the nucleus. The cell remains arrested in G1-phase for approximately 40 hr. (C, D) After treatment of a late G2-phase cell with 1 µM selinexor, it cell progresses normally to mitosis (M) and divides. One daughter cell is tracked and it enters G1-phase, progresses through a protracted early S-phase (red and green signal), transitions to solely green and dies after 21 hr 30 min. The data suggest S-phase progression is affected by selinexor treatment. Phase-contrast and fluorescent images were acquired at 1 frame per 10 min with a 20X PH2 0.70 NA lens. Bar = 10 µm. Movies 5 and 6. Please click here to view a larger version of this figure.
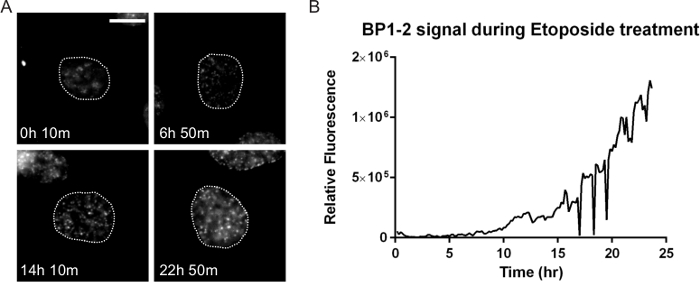
Figure 6. DNA Damage Dynamics after Drug Treatment. Many anti-cancer therapies result in DNA damage that can profoundly impact cell response and treatment success. HT1080 cells stably expressing both the double-strand DNA damage marker mCherry-BP1-2 and H2b-EGFP (not shown) were treated with 10 µM of the topoisomerase II drug etoposide and DNA damage was tracked. (A) The number and intensity of the foci increase after etoposide. There is initially a lag, indicating possible cell cycle effects consistent with the known mechanism of etoposide. By 22 hr 50 min this cell has accumulated high levels of damage. While not shown here, the fate of this cell can be determined by direct tracking. (B) A ROI corresponding to the nucleus obtained through the H2b-EGFP signal was tracked using particle tracking in ImageJ and the integrated BP1-2 mCherry signal was quantified and plotted over time. The lag in signal until approximately 10 hr is noted, followed by a persistent increase. Fluorescent images were acquired at 1 frame per 10 min with a 40X PH2 0.75 NA lens. Bar = 10 µm. Movie 7. Please click here to view a larger version of this figure.
Movie 1. Phase-contrast Time-lapse Microscopy of Wildtype MCF7 Cells after Anti-mitotic Drug Treatment. Wildtype MCF7 cells were treated with 500 nM Kinesin-5 inhibitor and imaged every 10 min for 96 hr using phase-contrast microscopy with a 20X PH2 0.70 NA lens. Prolonged mitotic arrest and exit from mitosis can be observed, as described in Figure 2. Please click here to download this file.
Movie 2. Phase-contrast Time-lapse Microscopy of p53-knockdown MCF7 Cells after Anti-mitotic Drug Treatment. MCF7 cells stably expressing a small hairpin RNA targeting p53 for degradation were treated with 500 nM Kinesin-5 inhibitor and imaged every 10 min for 96 hr using phase-contrast microscopy with a 20X PH2 0.70 NA lens. Prolonged mitotic arrest and multiple rounds of mitosis can be observed, as described in Figure 2. Please click here to download this file.
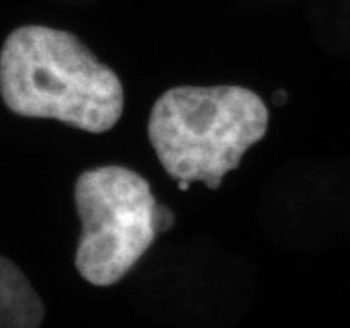
Movie 3. Fluorescent Time-lapse Microscopy of HeLa Cells after Low-dose Paclitaxel Treatment. HeLa cells stably expressing H2B-mCherry and β-tubulin-EGFP were treated with 1 nM paclitaxel. Chromosome attachment and segregation issues can be observed, as described in Figure 3. Images were acquired every 10 min with a 20X PH2 0.70 NA lens. Please click here to download this file.
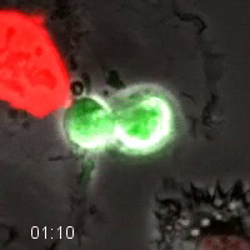
Movie 4. Fluorescent Cell Cycle Markers Allow for Direct Monitoring of Cell Cycle Progression. HT1080 cells expressing the FUCCI system were longitudinally tracked during time-lapse microscopy. The cell goes dim after exiting mitosis, and then becomes red as the cell progresses through G1-phase. As the cell enters S-phase, it becomes yellow as the green geminin probe increases and the red Cdt1 probe is degraded. The cell remains green as it progresses through S- and G2-phase. The green degrades as the cells enters anaphase. Quantification of this cell is shown in Figure 4. Images were acquired every 10 min with a 20X PH2 0.70 NA lens. Please click here to download this file.

Movie 5. HT1080 Cell Expressing Fluorescent Cell Cycle Markers after Treatment with a G1-phase Inhibitor. HT1080 cells expressing the FUCCI system were treated with 10 µM PD0332991, a Cdk4/6 inhibitor. The tracked cell progresses normally to mitosis and divides. One daughter is tracked, and remains red in G1 for the duration of the movie. Quantification is shown in Figure 5. Images were acquired every 10 min with a 20X PH2 0.70 NA lens. Please click here to download this file.
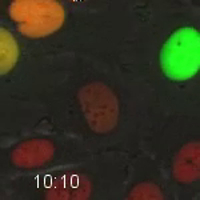
Movie 6. HT1080 Cell Expressing Fluorescent Cell Cycle Markers after Treatment with the Exportin-1 Inhibitor, Selinexor. HT1080 cells expressing the FUCCI system were treated with 1 µM selinexor. This late G2-phase cell was tracked through mitosis. A daughter cell then progresses through G1-phase (red) and enters S-phase (yellow). The cell progresses slowly through S-phases until it enters late-S/G2-phase and dies after 21 hr 30 min of treatment. Quantification is shown in Figure 5. Images were acquired every 10 min with a 20X PH2 0.70 NA lens. Please click here to download this file.
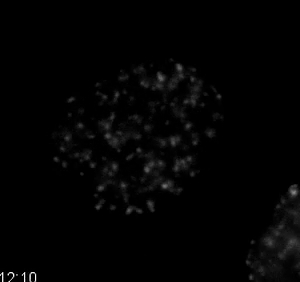
Movie 7. DNA Damage Dynamics after Treatment with a Topoisomerase II Inhibitor. HT1080 cells stably expressing the double-strand DNA damage marker mCherry-BP1-2 and H2B-EGFP were treated with 10 µM etoposide, a topoisomerase II inhibitor. The mCherry-BP1-2 is displayed in the movie. As treatment continues, the signal of mCherry-BP1-2 increases, indicating increased double-strand DNA damage. Quantification is shown in Figure 6. Images were acquired every 10 min with a 40X PH2 0.75 NA lens. Please click here to download this file.