The permeable membrane insert-based infection system protocol developed for the study of secreted bacterial factors is detailed in Figure 1. This system relies on the separation of bacteria and host cells via a porous membrane to assess the effects of secreted bacterial factors, in this case Streptolysin S (SLS), on host responses such as host membrane integrity, cellular viability, cellular signal transduction, and secreted host cell factors. Figure 2 provides representative Western Blot data demonstrating that this system may be used to assess changes in the activation of contact-independent host signaling proteins. Specifically, the representative data show significantly enhanced p38 MAPK activation in the presence of SLS-producing GAS strains. This system can also be applied to visualize the effects of secreted bacterial factors on host protein localization by immunofluorescence microscopy (Figure 3). The data show SLS-dependent activation of the key inflammatory mediator Nuclear Factor kappa B (NFκB), which translocates from the cytoplasm to the nucleus upon activation21. The results in Figures 2 and 3 indicate that both SLS-dependent inflammatory signaling responses do not require direct contact between the bacteria and host cells. As previous experiments have already demonstrated SLS-dependent p38 and NFκB activation in direct infection models21, similar levels of p38 or NFκB activation among the three GAS strains in the permeable membrane insert-based system would have indicated that the response required direct contact between the bacteria and host cells. Figure 4 demonstrates that this infection system may be applied to assess toxin-dependent changes in host cytotoxicity via ethidium homodimer and LDH release assays. This is evidenced by the significant increase in both membrane permeabilization and in the release of LDH from host cells exposed to GAS strains containing SLS compared to uninfected cells or cells exposed to the SLS-deficient strain. These changes in cytotoxicity are not immediate, as significant effects are not evident until 12 hr post infection in the case of membrane permeabilization and 16 hr in the case of LDH release. The representative data illustrate the importance of selecting appropriate time points and infection conditions for the evaluation of these host responses. In addition to allowing for the assessment of host signaling changes and cytotoxicity, the permeable membrane insert-based infection system is applicable to the study of metabolic changes such as accurate determination of host ATP levels by preventing bacterial contamination of host cell lysates (Figure 5). These data show a significant loss of keratinocyte ATP in response to GAS infection by 16 hours post infection, with enhanced ATP loss in the presence of SLS. These results are consistent with the observed toxin-dependent increases in host stress-response signaling and cytotoxicity.
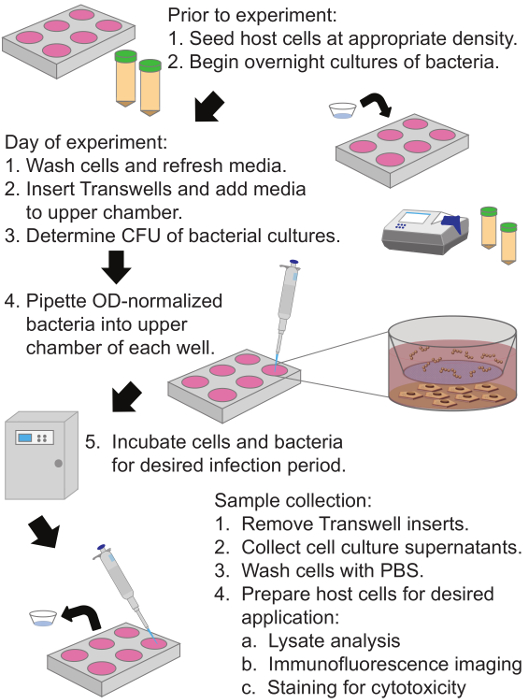
Figure 1: Diagram of Permeable Membrane Insert-Based Infection System Protocol to Assess the Effects of Secreted Bacterial Factors on Host Cells. Human keratinocytes are plated in the bottom compartment and grown to 90% confluence. A collagen coated membrane with 0.4 µm pores separates the upper and lower chambers, and bacteria are added to the upper chamber of the permeable membrane insert system for the desired infection period. Cell culture supernatants and host cells may be collected following the infection period and utilized for a variety of analyses. Please click here to view a larger version of this figure.
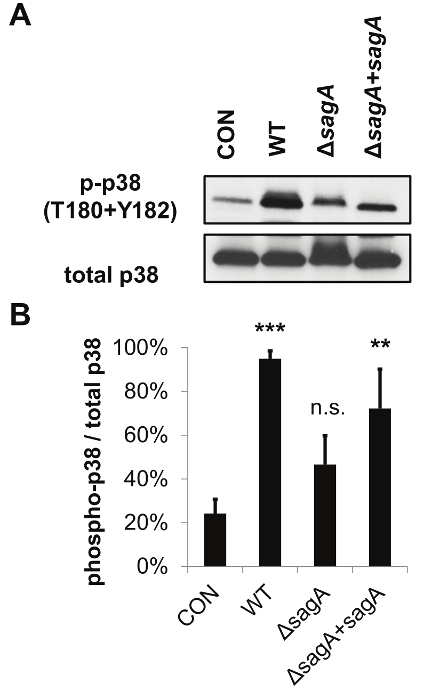
Figure 2: The Permeable Membrane Insert-Based Infection System can be Utilized for Host Cell Lysate Analysis by SDS-PAGE and Western Blotting. The representative data demonstrate that SLS enhances activation of the p38 MAPK pathway in infected keratinocytes. (A) HaCaTs were infected with GAS for 7 hr via the permeable membrane insert infection system (at MOI = 10) and lysates were assessed for activation of p38. (B) Densitometry from three independent Western Blots was performed to quantify the relative activation of p38 in response to GAS'infection. Averages from three biological replicates are shown, with error bars representing standard deviation. Relative activation of p38 is represented as phosphorylated/total protein level. Statistical significance was determined compared to uninfected cells. The overall p-value was determined by ANOVA (p = 0.0063). Dunnett's tests were performed post hoc to compare each condition to the corresponding uninfected control mean. *, p = 0.01-0.05; **, p = 0.001-0.01; ***, p = 0.0001-0.001; ****, p <0.0001. This figure has been modified from Flaherty et al. 201521. Please click here to view a larger version of this figure.
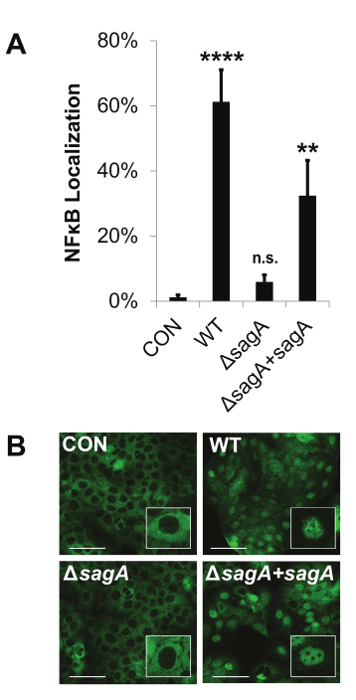
Figure 3: The Permeable Membrane Insert-Based Infection System Allows for the Visualization of Host Signaling Changes by Immunofluorescence Microscopy. The representative data show that Streptolysin S enhances pro-inflammatory signaling through activation of NFκB. HaCaT human keratinocytes were infected with GAS at an MOI of 10 for 8 hr using the permeable membrane insert-based infection system. (A and B) Nuclear localization of NFκB was assessed by immunofluorescence imaging. The percentage of nuclear localized cells was calculated by counting the number of cells in which NFκB had translocated from the cytoplasm to the nucleus for a given field and dividing that number by the total number of cells for the same field. Scale bars indicate 100 µm. (A) The average of three biological replicates are represented for each condition with error bars representing standard deviation. The overall p-value was determined by ANOVA; p <0.0001. Dunnett's tests were performed to compare each condition to the corresponding uninfected control condition. *, p = 0.01-0.05; **, p = 0.001-0.01; ***, p = 0.0001-0.001; ****, p <0.0001. This figure has been modified from Flaherty et al. 201521. Please click here to view a larger version of this figure.
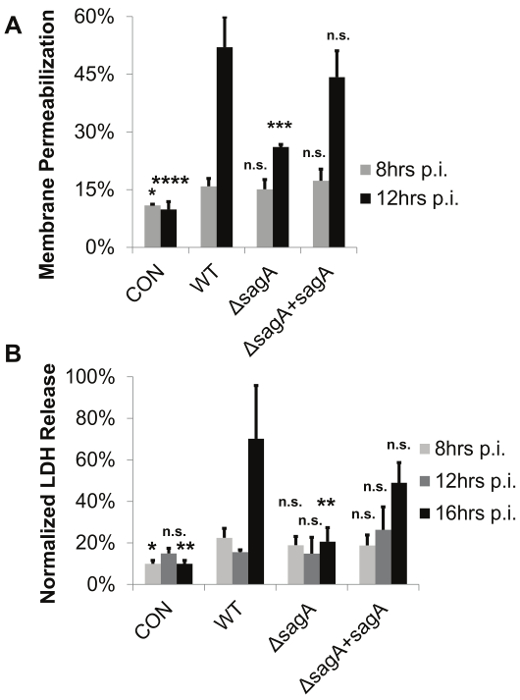
Figure 4: The Permeable Membrane Insert-Based Infection System Allows for the Determination of Bacterial-Mediated Membrane Permeabilization and Cytotoxicity in the Absence of Direct Contact between Bacteria and Host Cells. The representative data demonstrate that keratinocyte viability decreases in the presence of active SLS toxin. GAS–induced cell death was assessed in HaCaT cells in the presence of WT, SLS-deficient or sagA complemented GAS. Keratinocytes were exposed to GAS using the permeable membrane insert-based infection system for 8-16 hr at an MOI of 10. (A) Viability was assessed by ethidium homodimer assay or (B) LDH release assay. In both panels, 3 replicates are averaged and error bars represent standard deviation. Significance for each time point was determined by ANOVA (A) 8 hr, p = 0.0241; 12 hr, p <0.0001 (B) 8 hr, p = 0.0287; 12 hr, p = 0.1977; 16 hr, p = 0.0031. Dunnett's tests were performed to compare means from each condition to the wild-type infection for the corresponding time point. *, p = 0.01-0.05; **, p = 0.001-0.01; ***, p = 0.0001-0.001; ****, p <0.0001. This figure has been modified from Flaherty et al. 201521. Please click here to view a larger version of this figure.
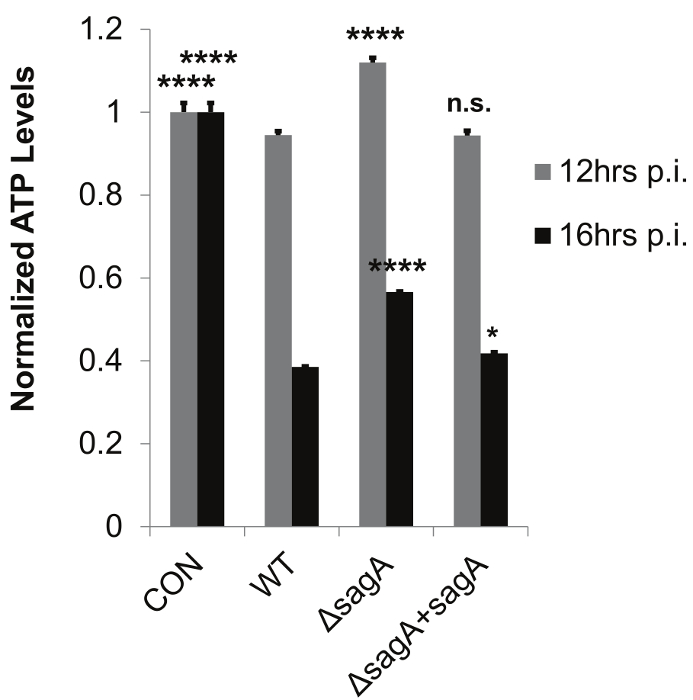
Figure 5: The Permeable Membrane Insert-Based Infection System Allows for Accurate Determination of Host ATP Levels by Preventing Bacterial Contamination of Host Cell Lysates. The representative data demonstrate SLS-dependent loss of ATP during Group A Streptococcal infection. HaCaT cells were infected with GAS for 8-16 hr using the permeable membrane insert-based infection system. Technical replicates (n = 3) from one representative biological replicate (2 x 106 cells per sample) were averaged for each condition, with error bars representing standard deviation. The overall p-values were determined by ANOVA (12 hr, p <0.0001; 16 hr, p <0.0001). Dunnett's tests were performed to compare each condition with wild-type infection for the corresponding time point. *, p = 0.01-0.05; **, p = 0.001-0.01; ***, p = 0.0001-0.001; ****, p <0.0001. This figure has been modified from Flaherty et al. 201521. Please click here to view a larger version of this figure.