The photostimulation can be performed simultaneously along with continuous confocal scanning microscopy. The photostimulation can start at any pre-defined time slot in the time-lapse confocal microscopy sequence. The confocal microscopy can monitor cellular molecules by fluorescent imaging. The molecular responses to photostimulation and other dynamics can be identified in this way. Theoretically, if ERK is activated, it will be phosphorylated move from the cytoplasm to cell nucleus27. The specific cell fate can be regulated by the certain patterns of this ERK signal28. In recent studies, optical modulation based on optogentics has provided a high precise control of the ERK signal in duration and magnitude and revealed that perturbed ERK signal transmission dynamics drives improper proliferation in cancer cells7,8. Here, we demonstrate the ERK2 translocation into nucleus after treating cell with a short flash of femtosecond laser by using the method presented in this protocol. As shown in Figure 5A, ERK2-GFP fluorescence reaches the maximum after several minutes of femtosecond laser simulation. The ERK2 molecules will be dephosphorylated after activating downstream substrates in the nucleus, and then the ERK2 comes back to the cytoplasm indicated by decreasing of nuclear GFP fluorescence. ERK2 can be activated for multiple times by multiple photostimulations (Figure 5B). Therefore, it is able to manipulate the ERK2 signal pattern precisely by controlling interval time between multiple stimuli. In addition, ERK2 can be activated in adjacent cells around the stimulated cell occasionally (Figure 5C). This observation indicates that some diffusible molecules may be released by the cell treated with femtosecond laser to activate ERK2 in the adjacent cells. Phosphorylation of ERK2 downstream protein eIF4E can be confirmed and visualized by immunofluorescence microscopy (Figure 5D). This result indicates that femtosecond laser stimulation can successfully activate ERK signaling pathway. More detailed results are in Wang S., et al.22.
Mitochondrial oxidative flashes (mitoflashes) are oxidative bursts in mitochondria that root from complex mitochondrial molecular dynamics. In last past decade, mitoflashes are realized to be an elemental mitochondria signaling event and take an important part in multitudinous cell functions29,30,31. Traditionally, mitoflashes are usually observed by chance when treating cells with chemicals to provide indirect stress to mitochondria29,30. By implementing this photostimulation scheme, we achieve a controllable and precise manner to excite mitoflashes at single mitochondrial tubular level. The successful mitoflash excitation is shown in Figure 6A. Interestingly, the properties of mitoflashes such as pulse peak, width and response durations32 are closely related to the femtosecond laser power. More detailed quantitative analysis of mitoflashes excited by femtosecond laser stimulation is in Wang S., et al.32. This photostimulation to mitochondria also shows varieties of mitochondrial molecular dynamics, including fragmentation and restoration of mitochondrial morphology (Figure 6B), and oscillation of mitochondrial membrane potential (Figure 6C). Similar to photo-activated mitoflashes, these mitochondria events have different performance with different power intensities of photostimulation. It is different from activation of ERK signaling pathway. The influence of femtosecond laser is highly restricted on a single mitochondrial tube. More detailed results are in Wang Y., et al.24 and Shi F., et al.25.
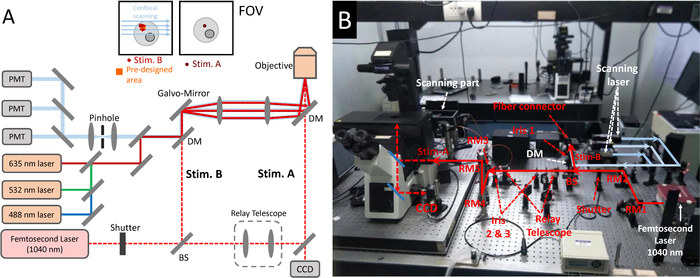
Figure 1: The photostimulation scheme established on a femtosecond laser coupling into a confocal microscope. (A) Optical paths of (B) the photostimulation and confocal imaging system. The femtosecond laser is at first split by a 50/50 beam splitter into two beams. The transmission is expanded by a relay telescope and then reflected into the objective to form Stim-A. The reflection beam is aligned through the microscope scanning system to form Stim-B. A CCD camera is used to provide a bright-felid imaging to monitor cells and focus of femtosecond laser in Stim-A mode. Stim-A = a fixed focus in the center of FOV; Stim-B = a special scanning frame at pre-designed area. DM = dichroic mirror, BS = beam splitter, RM = reflective mirror. The wavelength of confocal scanning laser is 488 nm/532 nm/635 nm, and the typical collection wavelength interval of fluorescence is <560 nm/560-625 nm/> 625 nm. The fiber femtosecond laser (1,040 nm, 120 fs, 50 MHz, 1 W) can be replaced by a Ti = Sapphire laser (810 nm, 80 MHz, 65 fs, 1 W) or other commercial femtosecond laser oscillators. Please click here to view a larger version of this figure.
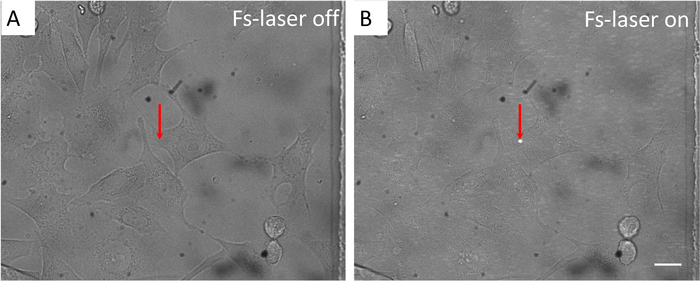
Figure 2: In Stim-A mode, the localization and size of femtosecond laser focus on the target cell at confocal imaging plane under bright-felid imaging. (A) Femtosecond laser is blocked by the shutter. (B) Femtosecond laser is on. Arrow: the reference arrow is set at the center of FOV to confirm that the femtosecond laser focus locates the correct position. Scale bar = 10 µm. Please click here to view a larger version of this figure.
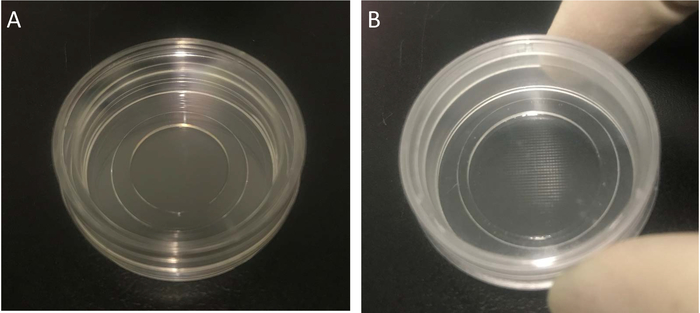
Figure 3: The Petri dish used for cell culture, transfection and photostimulation experiments. (A) The 35 mm Petri dish with a 15 mm diameter and 0.17 mm thickness glass bottom. (B) The 35 mm Petri dish with a glass bottom and an imprinted 500 µm cell location grid. Please click here to view a larger version of this figure.

Figure 4: The CO2 incubation system. (A) The CO2 incubator stage and (B) the control panel. Please click here to view a larger version of this figure.
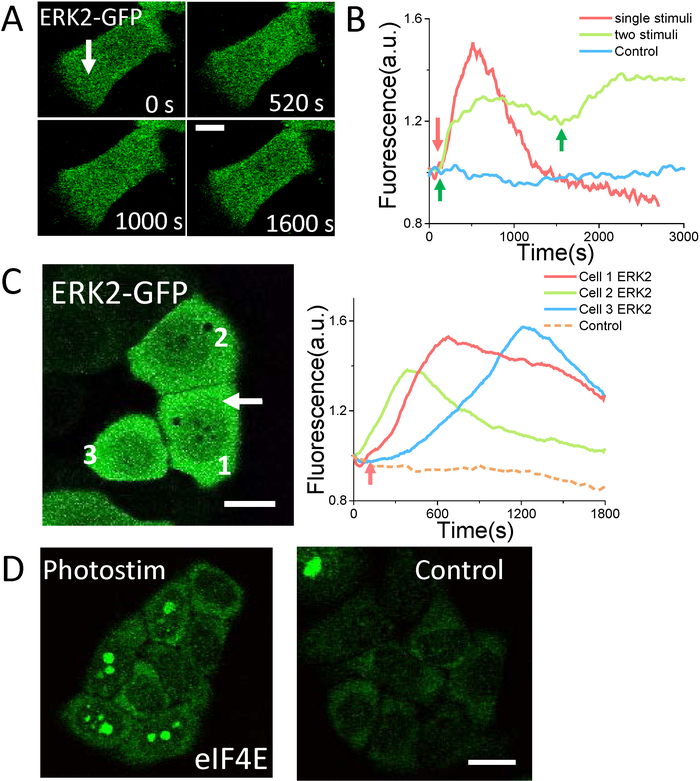
Figure 5: ERK photoactivation by using Stim-A mode. (A) Single photo stimulus (1,040 nm, 40 mW, 0.1 s) induces ERK2-GFP translocation into nucleus and then back to cytoplasm. (B) ERK2 signal pattern mediated by single femtosecond laser exposure (red arrow, 1,040 nm, 40 mW, 0.1 s) and two photostimulations (green arrows, 810 nm, 30 mW, 0.1 s). (C) ERK activation in surrounding cells by single short femtosecond laser exposure in target cell (white arrow, 810 nm, 24 mW, 0.2 s). (D) The Immunofluorescence of eIF4E-P shows a significant increase 24 h after single photosimulation (810 nm, 25 mW, 0.2 s). Scale bar = 10 µm (A), and 20 µm (B, C). The fluorescence of ERK2-GFP and eIF4E-P is excited by 488 nm excitation laser and collected in <560 nm channel. Please click here to view a larger version of this figure.
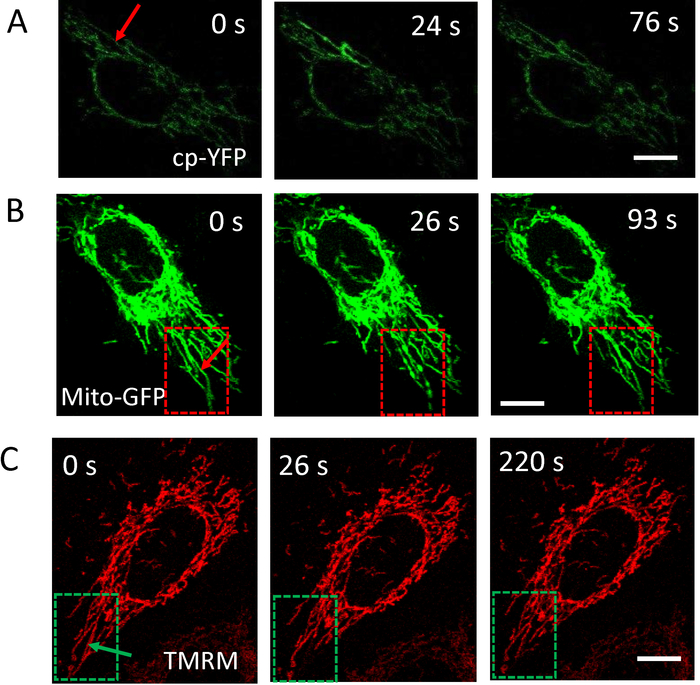
Figure 6: Multiple mitochondria events induced by photostimulation under Stim-A mode. (A) Excitation of mitoflash on the stimulated mitochondrial tubule by single stimulus (810 nm, 16 mW, 0.1 s). (B) Mitochondria morphological fragmentation and restoration induced by single photostimulation (1,040 nm, 30 mW, 0.1 s). (C) Oscillation of mitochondrial membrane potential by single femtosecond laser stimulation (1040 nm, 20 mW, 0.1 s). Arrows in (A) (B) and (C) indicate the position of photostimulations.Scale bar = 10 µm. The fluorescence of Mito-GFP or mt-cpYFP is excited by 488 nm excitation laser and collected in <560 nm channel. The fluorescence of TMRM is excited by 532 nm excitation laser and collected in 560-625 nm channel. Please click here to view a larger version of this figure.
| 0.05 s | 0.1 s | 0.2 s | 0.5 s | |
| 810 nm, 65 fs, 80 MHz | 20 – 65 mW | 10 – 60 mW | 5 – 50 mW | 5 – 40 mW |
| 1040 nm, 120 fs, 50 MHz | 30 – 100 mW | 20 – 80 mW | 15 – 70 mW | 10 – 60 mW |
Table 1: Recommended stimulation duration and average power of femtosecond laser in Stim-A mode.
| 0.05 s | 0.1 s | 0.2 s | 0.5 s | |
| 810 nm, 65 fs, 80 MHz | 25 – 40 mW | 20 – 30 mW | 15 – 25 mW | 10 – 25 mW |
| 1040 nm, 120 fs, 50 MHz | 40 – 60 mW | 30 – 50 mW | 25 – 40 mW | 20 – 30 mW |
Table 2: Recommended stimulation duration and average power of femtosecond laser in Stim-A mode at 2 x 2-3 x 3 µm2 in cell.