We performed relative quantification of 3 proteins from 10 samples, 5 samples from each group of patients with abnormalities in the brain. These proteins included Apolipoprotein A-I (APOA-I), Vimentin (VIM) and Nicotinamide phosphoribosyltransferase (NAMPT) which are known to perform diverse roles in the brain cells. Post-run analysis of the data was performed using Skyline-daily (Ver 20.2.1.286). A total of 10 peptides corresponding to 3 proteins were monitored. These included 3 peptides for APOA-I, 4 peptides for VIM and 3 peptides for NAMPT. The total number of transitions from these 10 peptides amounted to 57. The samples were grouped into either of the two groups depending on the condition they belonged to. Using the group comparisons feature of skyline, the peak abundances of these peptides were compared, and relative quantification values were calculated (Figure 3).
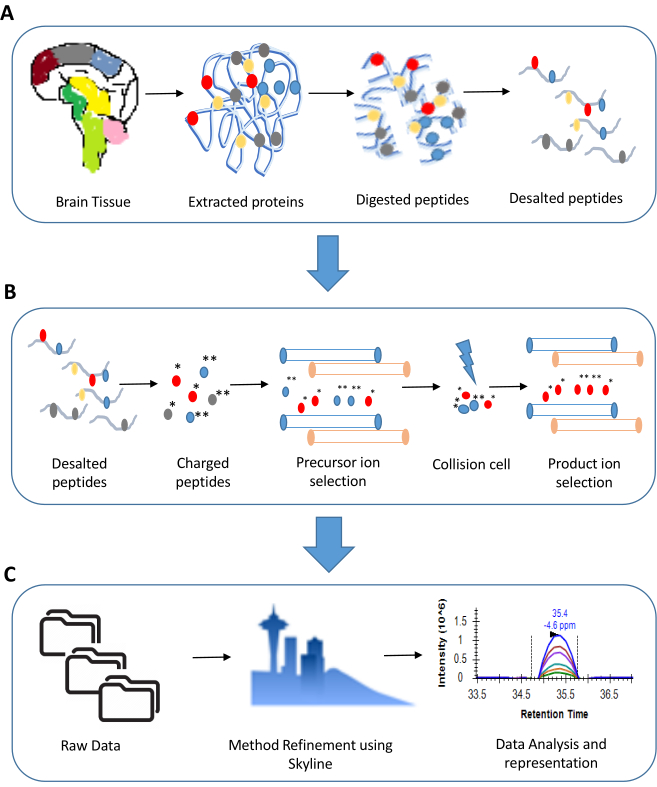
Figure 1: An overview of steps involved in a Multiple Reaction Monitoring experiment. A. Sample preparation for a typical proteomics experiment involves extraction of proteins (for illustration we have shown tissue sample) followed by digestion using trypsin. The digested peptides are ultimately desalted and made LC-MS ready. B. The steps involved in an MRM experiment include precursor and product ion selection based on their m/z values. Only the transitions showing good response are considered for analysis. C. The data analysis in an MRM experiment includes a detailed examination of peak shapes and peak areas. This is ultimately followed by statistical analysis of the results. Please click here to view a larger version of this figure.
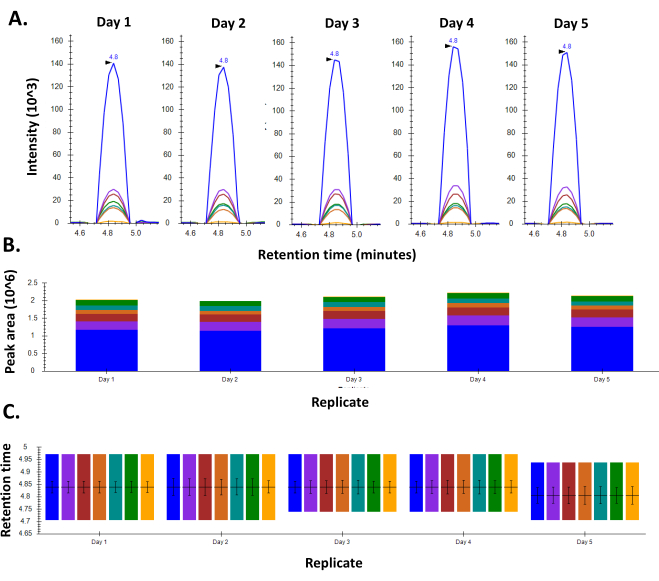
Figure 2: Consistency in response for BSA using an optimized MRM method. A. Chromatogram for a representative peptide of BSA shows consistent peak shape and intensity throughout the five days the experiment was performed. B. Retention time consistency observed for the peptide on all the five days of the experiment C. Peak areas for the peptide as seen over the course of five days in the week. Please click here to view a larger version of this figure.
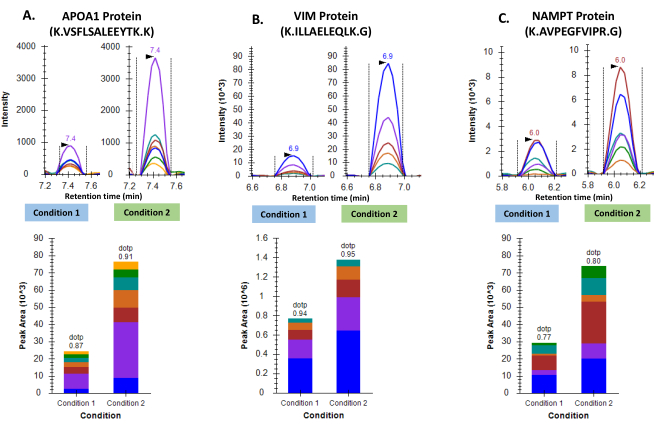
Figure 3: Differential regulation of three proteins in two groups of GBM tumor samples. A. Representative chromatograms for Apolipoprotein A-I and cumulative peak area as seen following inter-group comparison. B. Representative chromatograms for Vimentin and cumulative peak area as seen following inter-group comparison. C. Representative chromatograms for Nicotinamide phosphoribosyltransferase and cumulative peak area as seen following inter-group comparison. Please click here to view a larger version of this figure.
Table S1: Details of 10-minute LC gradient to be used for all samples. Please click here to download this Table.
Table S2: Parameter settings for the ion source. Please click here to download this Table.
Table S3: Parameter settings for MRM method. Please click here to download this Table.