Using this protocol, partially purified proteoliposomes can be obtained in a short time. Representative results are shown in Figure 2A. Twenty five GPCRs of Class A, B, and C were successfully synthesized using the bilayer-dialysis method (small scale) and partially purified by centrifugation and buffer wash. Although the amount of synthesized proteins varies according to the type of protein, 50 to 400 µg of membrane proteins usually can be synthesized per reaction when large dialysis cups are used. Several milligrams of membrane proteins can be easily produced by increasing the number of reactions, due to the high scalability of wheat cell-free system. A pre-test using a small dialysis cup is sufficient to determine the production efficacy of the target protein in bilayer-dialysis method. According to the obtained productivity, the amount of the target protein to be produced using large dialysis cups can be estimated.
This protocol is suitable for expression of membrane proteins, particularly for those with multiple transmembrane helices. In most cases, membrane proteins with three or more transmembrane helices are easily incorporated into proteoliposomes after synthesis (Figure 2B), which makes a good productivity of proteoliposomes. Single-transmembrane-helix proteins are usually synthesized smoothly; however, they hardly integrate into liposomes due to the small hydrophobic region. Regarding proteins with two transmembrane helices, whether or not they are anchored to liposomes is dependent on the way their transmembrane helices are exposed.
Synthesized proteoliposomes are collected by simple centrifugation, and partially purified with a washing buffer, which greatly shortens the purification process of membrane proteins. Although both biological membranes and membrane proteins have been removed from wheat germ extracts beforehand, small amounts of wheat proteins are sometimes co-precipitated by binding to liposomes or membrane proteins synthesized (Figure 2A). Such protein contaminants are difficult to remove by simple centrifugation and buffer wash. When a highly purified membrane protein is required, it is necessary to solubilize the partially purified proteoliposomes with a surfactant and purify them by column chromatography.
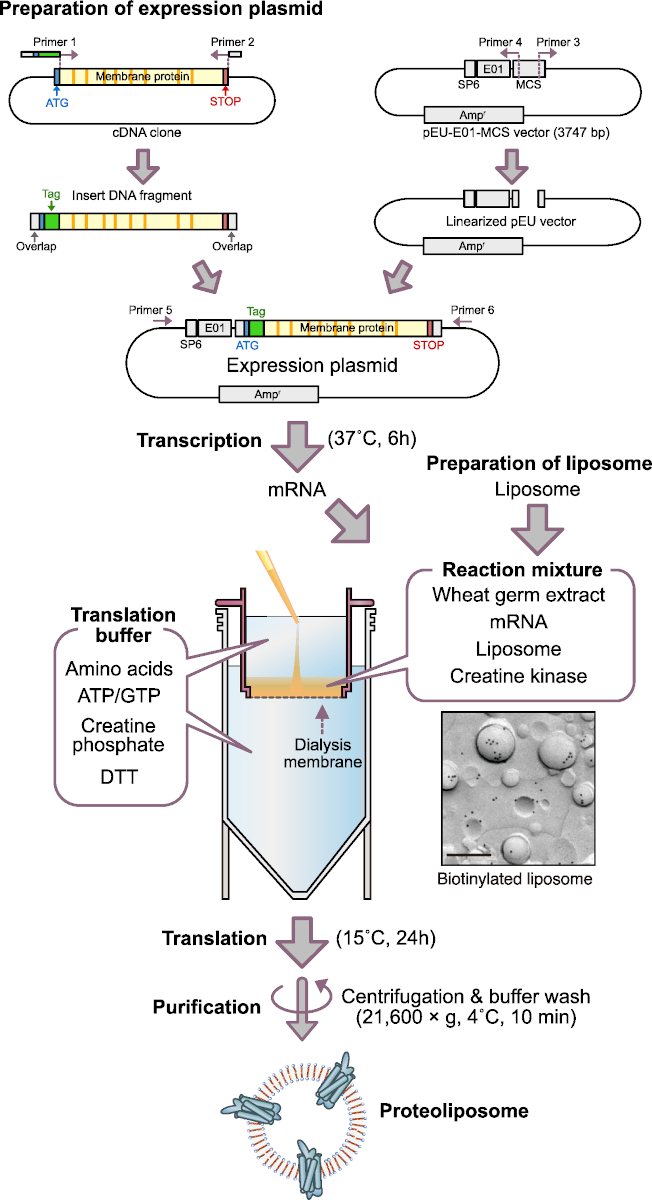
Figure 1: Scheme of cell-free proteoliposome production. SP6, SP6 promoter sequence; E01, E01 translation enhancer sequence; Ampr, ampicillin resistance gene; DTT, dithiothreitol. Electron micrograph shows immunogold labeling of biotinylated lipid containing liposome. Bar, 0.2 μm. This electron micrograph image was from Figure 1D in Takeda et al., 201545. Please click here to view a larger version of this figure.
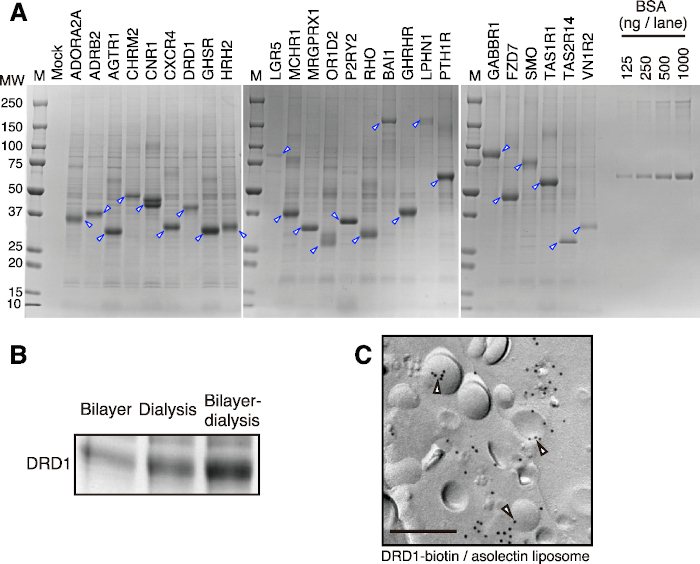
Figure 2: Representative results of proteoliposome production by bilayer-dialysis method. (A) SDS-PAGE image of cell-free synthesized GPCRs. Twenty-five selected GPCRs were synthesized by the bilayer-dialysis method. Proteoliposomes were partially purified and applied to SDS-PAGE and CBB staining. Arrowheads indicate target GPCRs. (B) Comparison of membrane protein productions between different translation methods. Dopamine receptor D1 (DRD1) protein was synthesized by each method in the same ratio of wheat germ extract, liposomes, and mRNA, respectively. DRD1 proteoliposome was partially purified by centrifugation, and subjected to SDS-PAGE and CBB staining. (C) Immunogold labeling of DRD1-biotin/liposome complex. DRD1 was enzymatically biotinylated by BirA biotin ligase. Bar, 0.2 μm. Blank arrowheads indicate DRD1-biotin on liposomes. This figure was modified from Figure 1 in Takeda et al., 201545. Please click here to view a larger version of this figure.
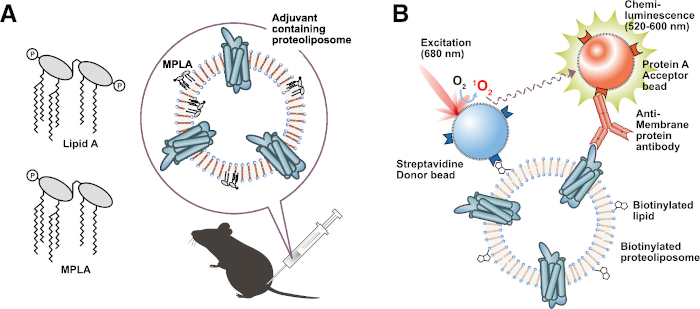
Figure 3: Application of functional proteoliposomes. (A) Immunization of adjuvant lipid-containing proteoliposome. (B) Biotinylated liposome-based interaction assay (BiLIA). Interaction between membrane protein and anti-membrane protein antibody was detected by AlphaScreen. Please click here to view a larger version of this figure.