Following the protocol, a 48 h timepoint was chosen to generate representative results. The creation of the initial wound in surplus tissue from elective cosmetic surgery can be seen in Figure 2A where the excised wound is clearly visible. Haematoxylin and eosin staining confirms that this has generated a full thickness wound (Figure 2B). After 48 h, partial closure of the wound is visible under the light microscope (Figure 2C). Histological staining reveals the epithelial tongue that is progressing to heal the wound (Figure 2D), demonstrating that the ex vivo wound healing model is a valid proxy for in vivo wound healing.
After sectioning and staining with haematoxylin and eosin, the healing wound was visualized on the laser capture microdissection system and the wound area selected (Figure 3A). This area was completely excised using this method as can be seen in Figure 3B. RNA quality and purity was reasonable (analyzed by RNA integrity number (RIN); Figure 3C) – poor quality amplified RNA would have many peaks and troughs indicating multiple degradation products. Isolation of small tissue sections may yield RNA that is of a very low concentration that makes valid interpretation of qPCR difficult. Figure 3D demonstrates the variation in RNA concentration to be expected using this technique, with a range of 2.00 to 6.15 ng/mL. Importantly, even dilute samples were able to give robust CT values for both housekeeper (GAPDH) and skin-specific genes of interest (keratin 17; KRT17; Figure 3E), confirming the suitability of the technique for comparative transcriptomic studies.
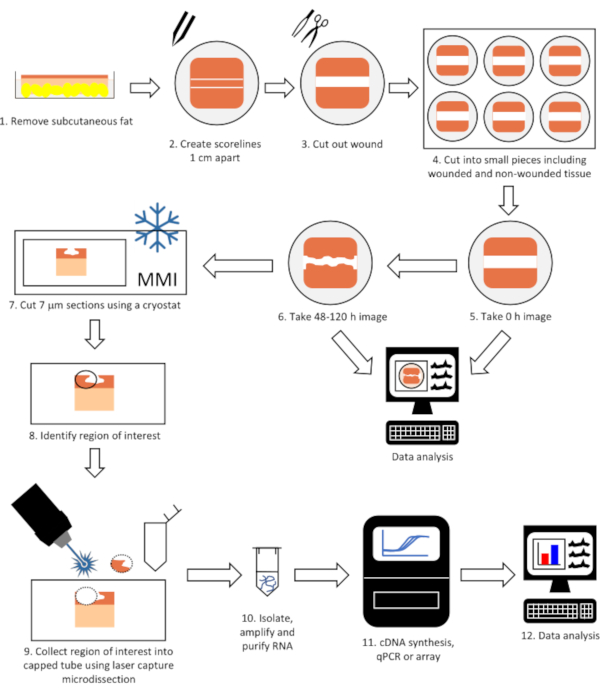
Figure 1: Flow diagram of the complete technique to perform gene expression analysis on laser microdissected tissue from wounded skin. Tissue is wounded, allowed to heal in a tissue incubator and imaged (steps 1-6) before being cut into 7 μm sections using a cryostat (step 7). The region of interest (e.g. epithelial tongue) is identified and collected using laser capture microdissection (steps 8-9) and RNA isolated, purified and gene expression determined (steps 10-12). Please click here to view a larger version of this figure.
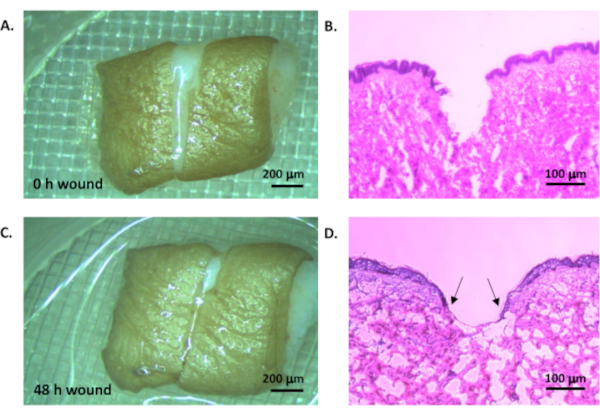
Figure 2: Ex vivo wound model. Human tissue was wounded by creating two parallel cuts and removing the tissue between to leave a uniform wounded area (A, light microscope, scale bar = 200 µm; B, haemoatoxylin and eosin staining, scale bar = 100 µm). The tissue was cultured in a standard tissue culture incubator at 37°C in 5% CO2 in air for 48 h (C, light microscope, scale bar = 200 µm; D, haematoxylin and eosin staining, scale bar = 100 µm). Arrow heads indicate epithelial tongue. Please click here to view a larger version of this figure.
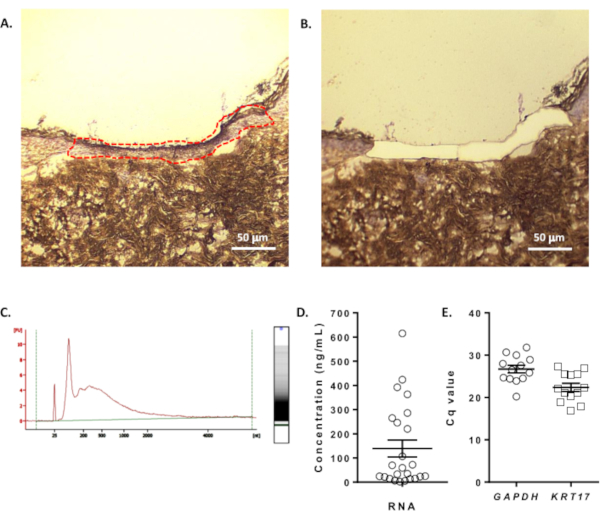
Figure 3: Laser capture microdissection and gene expression. A. The region of interest (in this case, healed tissue) was identified using haematoxylin staining and collected using laser capture microdissection. B. The same region imaged after microdissection. Scale bars = 50 µm. C. Representative electropherogram of RNA that was isolated, amplified and quantified from the laser microdissected tissue. D. RNA concentration from collected tissue (n=24 samples). E. Reproducible detection of GAPDH and KRT17 expression using qPCR (n=13 samples, mean ± SEM). Please click here to view a larger version of this figure.