Quality control analysis of the single-nucleus cDNA library preparation and snRNA-seq
Representative results describe sequencing results of 10,000 captured nuclei in a single pool with a 25,000 reads/nucleus gene expression library and a 5,000 reads/nucleus hashtag library. Figure 3B illustrates the quality control results of the 1st strand cDNA, gene expression (GEX) library, and HTO library, which were checked with Bioanalyzer. The HTO-derived cDNAs are expected to be smaller than 180 bp, whereas mRNA-derived cDNAs are larger than 300 bp. A high-quality GEX library can be detected as a broad peak from 300 to 1,000 bp, and the HTO library is detected as a specific peak of 194 bp. Cell Ranger was used for demultiplexing, fastq file generation, and read alignment by default setting. Seurat R package19 was subsequently used for quality control and downstream analyses.
Demultiplexing of the snRNA-seq data was performed by identifying HTOs using the Seurat built-in demultiplexing strategy. The demultiplexing ability for each HTO was first visualized with HTO expression files (Supplemental Figure S1). In the heatmap of Figure 4A, singlets are detected as nuclei with specific HTO expression, while doublets and negatives show nonspecific expression of multiple or no HTOs. Of note, approximately 33% of the nuclei were detected as negatives using a 10 min HTO antibody incubation approach. Prolongation of the incubation time (step 6.3) from 10 min to 30 min in a subsequent experiment revealed a remarkable decrease in negatively labeled nuclei (Supplemental Figure S2). These findings indicate that prolonging antibody incubation time may improve hashtag efficiency.
Violin plots in Figure 4B-D demonstrate the number of genes (nFeature_RNA), number of unique molecular identifiers (UMI) (nCount_RNA), and the percentage of mitochondrial counts (percent.mt) within the snRNA-seq dataset to identify outliers and low-quality nuclei. Nuclei were only included in downstream analysis when the following criteria were met: i) nFeature_RNA > 500 and nCount_RNA < 20,000; ii) percent.mt < 5%; iii) the individual nucleus showed clear expression of a single HTO. Gene expression counts were normalized using the default method in Seurat: 875 (2.71%) genes were detected as highly variable genes (Figure 4E). snRNA-seq GEX was scaled, was performed and the elbow plot was used to assess the inclusion of principal components that would be used for downstream analyses (Figure 4F). In total, 18 PCs were included. Clustering was performed with a resolution of 0.4.
The nuclei were clustered, and dimension reduction (UMAP) was performed for visualization of the 12 individual clusters (Figure 5A). The median raw gene count per cluster varies between 991.5 and 4,586 (Supplemental Figure S3A, B). Visualizing the division of the HTO antibodies within the UMAP reveals a clear distribution of ganglia, indicating that all clusters are presented in each ganglion (Figure 5B). To validate the accuracy of HTO sample segregation, the expression of X Inactive Specific Transcript (Xist, expressed in the inactive female X chromosome) was assessed to identify the male samples and the female samples (Figure 5C). Xist expression was in accordance with the hashtag labeling, showing that HTO 1-4 labeled samples were female samples, and HTO 5-8 labeled samples were male samples. This suggests that the curated HTO labeling is highly specific.
To further verify the quality and resolution of the sequencing data with the present method, some key transcripts of sympathetic neurons were first examined. The results show the presence of sympathetic neurons that highly express Th, Dbh, and Snap25 in clusters 5 and 7 (Figure 5D-F). Satellite glial cells were detected with the expression of S100b in clusters 0-3 (Figure 5G)20. Endothelial cells were detected in cluster 4 with a high expression of Pecam1 (Figure 5H) and stromal cells in cluster 8 with a high expression of Acta2 (Figure 5I). These results support the successful nuclei capture of neuronal, satellite glial, endothelial, and stromal cells of the sympathetic ganglion using snRNA-seq.
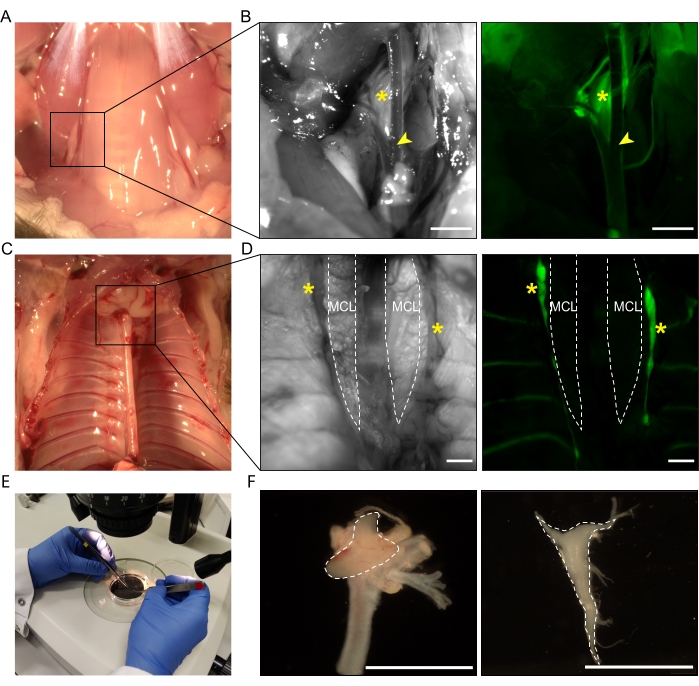
Figure 1: Dissection of adult mouse superior cervical ganglia and stellate ganglia. (A) Brightfield image of the location of the SCG. (B) To facilitate visualization, a Wnt1Cre;mT/mG mouse was used. Asterisks indicate the SCG (eGFP+), arrowheads indicate the bifurcation of the carotid artery. Left panel, phase contrast image; right panel, fluorescent image. (C) Brightfield image of the location of the StG. (D) Asterisks indicate the StG (eGFP+), dashed lines indicate the MCL. Left panel, phase contrast image; right panel, fluorescent image. (E) Dissected ganglia are transferred into a Petri dish separately for further cleaning under a stereomicroscope. (F) Left panel, the dissected SCG with the carotid artery still attached. Dashed outline indicates the SCG. Right panel,the dissected and cleaned StG has the shape of an inverted triangle, as indicated by the dashed outline. Scale bar = 1,000 µm. Abbreviations: SCG = superior cervical ganglia; StG = stellate ganglia; MCL = musculus colli longus; eGFP = enhanced green fluorescent protein. Please click here to view a larger version of this figure.
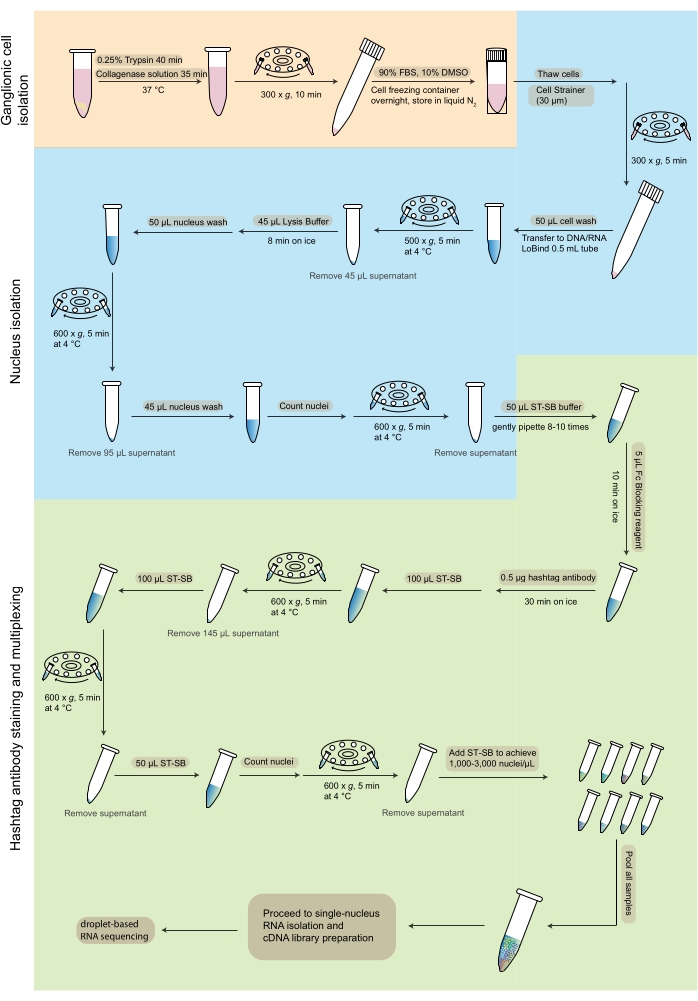
Figure 2: Workflow of sample preparation and hashtag staining-based multiplexing for snRNA-seq. The flowchart depicts the steps from the dissociation of ganglionic cells (orange), nucleus isolation (blue), and hashtag antibody staining and multiplexing (green) that are carried out for snRNA-seq. Abbreviations: FBS = fetal bovine serum; ST-SB = ST staining buffer; snRNA-seq = single-nucleus RNA sequencing. Please click here to view a larger version of this figure.
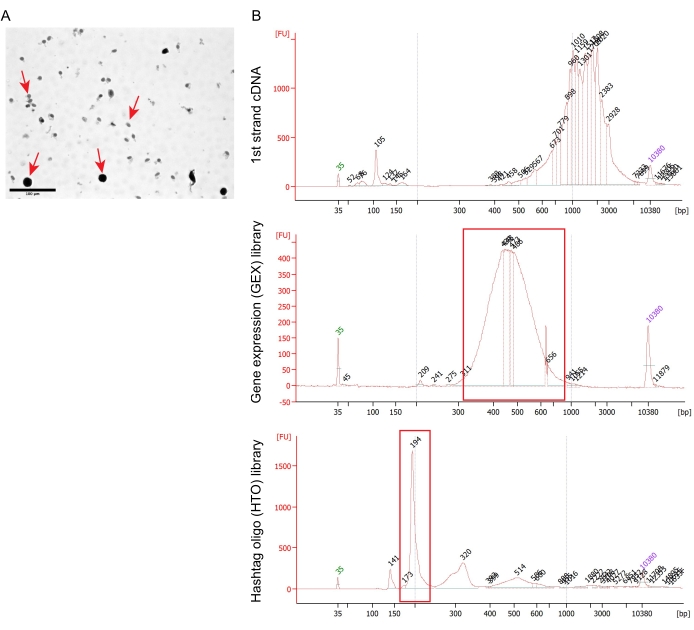
Figure 3: Quality control of nucleus isolation and gene expression library preparation. (A) Phase-contrast image of the HTO-stained nuclei. Nuclei are indicated with arrows. Scale bar = 100 µm. (B) Bioanalyzer results of 1st strand cDNA (top), GEX library (middle), and HTO library (bottom). Abbreviations: GEX = gene expression; HTO = hashtag oligo. Please click here to view a larger version of this figure.
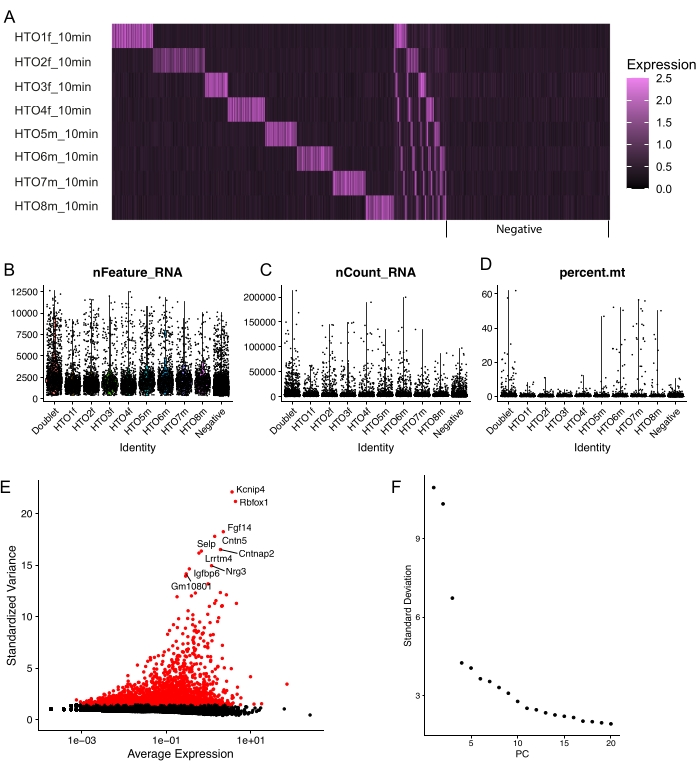
Figure 4: Quality control of the hashtag oligo labeling efficiency and quality control of snRNA-seq. (A) Heatmap of HTO staining achieved with an incubation time of 10 min with hashtag antibodies. (B–D) Violin plots of quality control metrics depicting the number of genes (nFeature_RNA, B); the number of UMIs (nCount_RNA, C); the percentage of mitochondrial counts (percent.mt, D). (E) Of the total 32,285 genes sequenced, 875 (2.71%) were identified as highly variable features as visualized in the scatter plot. (F) Elbow plot of PCs to determine inclusion of true signal used for clustering. Abbreviations: snRNA-seq = single-nucleus RNA sequencing; HTO = hashtag oligo; PCs = principal components. Please click here to view a larger version of this figure.
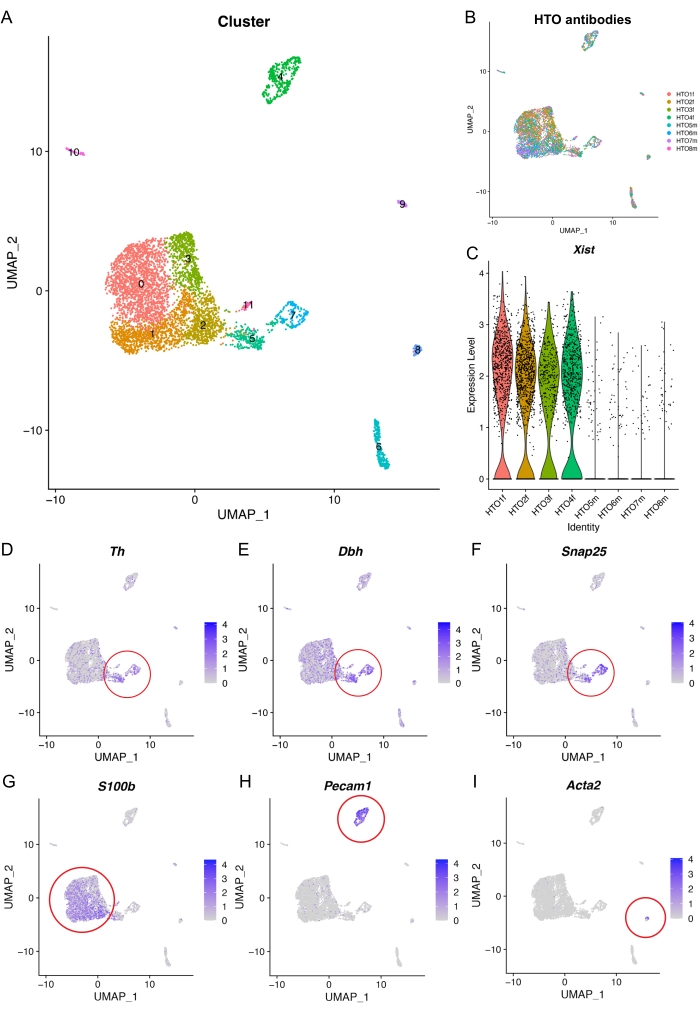
Figure 5: Representative results of the analysis of snRNA-seq. (A) UMAP plot of the clustered snRNA-seq dataset. (B) UMAP plot visualizing the distribution of the pooled HTO samples. (C) Violin plot validating female samples (high Xist expression) after demultiplexing. (D–I) UMAP plots displaying selected marker genes; the clusters highly express the corresponding genes which are indicated within the red circles: D–F, Sympathetic neurons (Th, Dbh, Snap25); (G) Satellite glial cells (S100b); (H) Endothelial cells (Pecam1); (I) Stromal cells (Acta2). Abbreviations: snRNA-seq = single-nucleus RNA sequencing; HTO = hashtag oligo; UMAP = uniform manifold approximation and projection. Please click here to view a larger version of this figure.
Supplemental Figure S1: Quality control metrics of a representative single-nucleus sequencing result. HTO expression profiles of individual samples visualizing the demultiplexing ability of the used hashtag antibodies. Abbreviation: HTO = hashtag oligo. Please click here to download this File.
Supplemental Figure S2: HTO expression heatmap of subsequent samples incubated with HTO antibodies for 30 min. Comparison of the number of negative-labeled nuclei after HTO demultiplexing with Figure 4A shows a marked improvement of nucleus labeling after prolonging the antibody incubation time. Abbreviation: HTO = hashtag oligo. Please click here to download this File.
Supplemental Figure S3: Median and quantiles of gene expression per cluster. (A) Median nonzero raw gene expression of each cluster. (B) Descriptive statistics of nonzero raw gene expression of each cluster. Q1: 25th percentile, Q3: 75th percentile. Please click here to download this File.