Following the protocol, the biofilms of Acinetobacter isolates, originally isolated from kitchen surfaces, were formed on a polystyrene 96-well plate, stained with crystal violet, and the dyes were solubilized in ethanol and measured for biofilm mass (Figure 1). The number of biofilms greatly varied depending on the strains ranging from OD 0.04 to 1.69 (Figure 1). Based on the criteria established by Stepanović et al.16, all of the isolates except for A. bouvetii formed biofilms. A. radioresistens formed a weak biofilm. A. junii and A. baumannii formed moderate biofilms, while A. pittii and A. ursingii formed strong biofilms.
To count viable cells in biofilms, the biofilms were scraped off using cell scrapers, vortexed at high speed, diluted, and spread on the Acinetobacter selective plates. After incubation, the number of colonies was counted to estimate the number of biofilm cells (Figure 2). All the isolates except for A. bouvetii had cells equivalent to 7-8 Log CFU in their biofilms. Consistent with the biofilm non-forming property shown by crystal violet assay, A. bouvetii had a much lower level of cell number at 4.4 Log CFU.
The bottom-surface attached biofilms were visualized using CLSM (Figure 3). A substantial amount of biofilm was found in A. junii, A. baumannii, and A. ursingii with distinct biofilm morphologies. While A. pittii was a strong biofilm former in crystal violet assay, it did not form much biofilm on the bottom surface.
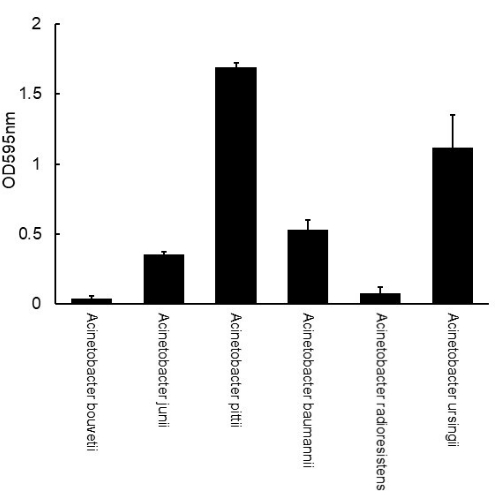
Figure 1: Measurement of biofilm mass of Acinetobacter formed on a polystyrene 96-well plate using crystal violet assay. The error bars represent the standard deviation from triplicate. Please click here to view a larger version of this figure.
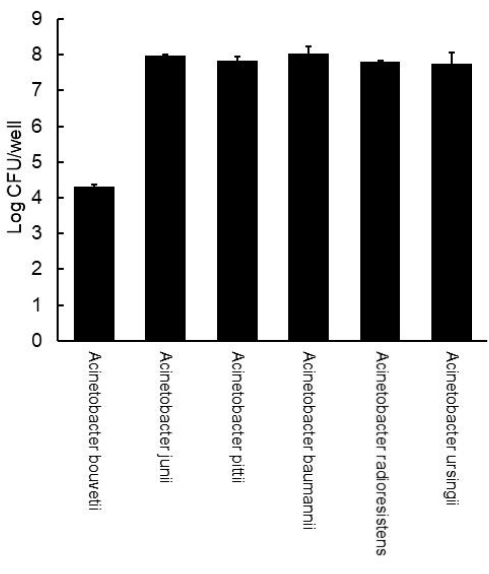
Figure 2: Viable counts of Acinetobacter biofilms formed on a polystyrene 12-well plate. The error bars represent the standard deviation from the duplicate. Please click here to view a larger version of this figure.
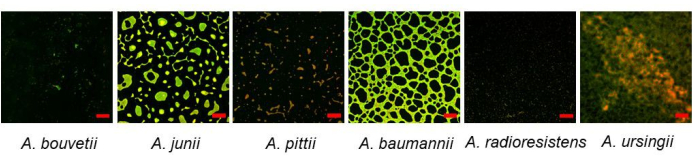
Figure 3: Bottom-surface attached Acinetobacter biofilms formed on a 96-well plate visualized by CLSM using SYTO 9 and propidium iodide dyes. Scale bars: 50 µm. Please click here to view a larger version of this figure.