1. Solution preparation
- Prepare 10X R1, 10X R2 and 10X R3 solutions according to the table.
R1
Chemicals MW(g/mol) mM (1X) 10X stock (g/L) NaCl 58.44 125 73.05 KCl 74.55 2.5 1.86 MgCl2 1 M stock 1 10 ml CaCl2·2H2O 147.02 2 2.94 NaH2PO4·H2O 137.99 1.25 1.72
R2
Chemical MW(g/mol) mM (1X) 10X stock (g/L) NaHCO3 84.01 25 21
R3
Chemicals MW(g/mol) mM (1X) 10X stock (g/L) NaCl 58.44 125 73.05 KCl 74.55 2.5 1.86 MgCl2 1 M stock 2 20 ml CaCl2·2H2O 147.02 2 2.94 HEPES 1 M stock 5 50 ml - Make 1 liter of mouse artificial cerebro-spinal fluid (mACSF) on the day of the experiment by mixing 100 ml of 10X R1 and 100 ml of 10X R2 in double distilled water (DDW) and add 1.8 g of dextrose (final 10 mM). This preparation method prevents calcium carbonate precipitation at high pH. The osmolarity of mACSF is around 310-315 mOsm/L. Adjust the osmolarity with dextrose or water if necessary. Aerate the solution with Carboxygen gas (95% oxygen and 5% carbon dioxide) for at least 30 minutes before adjusting the pH of the solution to 7.2-7.4.
- Make 1 liter of Ringer’s solution on the day of the experiment by mixing 100 ml of 10X R2 and 100 ml of 10X R3 in DDW and add 1.8 g of dextrose (10 mM). The osmolarity and pH of Ringer’s solution should be similar to those of the mACSF. Aerate the solution with Carboxygen gas.
- Prepare 4% low melting agarose (LMA) in either mACSF or Ringer’s solution. Aliquot the melted agarose in eppendorf tubes and store at 4°C till use. LMA, which is composed of purified polysaccharide, remains fluid at 37°C and solidifies quickly at temperature below 25°C. These properties make it ideal for embedding VNO and live tissue physiology. Other impure materials, such as agar, should not be the choice for live tissue experiment as uncharacterized components may interfere with neuronal response.
- Prepare pheromone stimuli in Ringer’s solution. For mouse urine, 1:100 dilution give rise to robust responses.
2. Preparation of VNO slice
- Before sacrificing the animal, put two tubes of LMA on a heat block and set the temperature to >60°C to melt the agarose. Once the gel liquefies, transfer the tubes to a 37°C heat block.
- Decapitate a G-CaMP2 mouse following CO2 euthanasia. Cut the mandible bones with scissors to remove the lower jaw. Peel off the ridged upper palate tissue to expose the nasal cavity (Figure 1A). Separate the jawbones by inserting a surgical blade between the two front incisors. Carefully remove the jawbones to expose VNO. At this point, the whole VNO can be lifted up from the nasal cavity by holding on the tail bone (Figure 1B). Transfer the VNO to cold oxygenated mACSF solution immediately.
- Under dissection scope, separate the two VNOs by using the tip of #5 forceps gently slide along the wall of septal bone (Figure 1C). Peel away the vomer bone that encases the VNO tissue. Gently lift the entire VNO tissue from the bone cavity. Extreme care should be taken in this step not to damage the neuroepithelium. Small bone fragments left on the tissue surface must be removed completely before embedding. Fragments left on the tissue may be caught by the cutting blade to pull the tissue out of the agarose block.
- Use the forceps to hold the posterior end of the VNO tissue and gently submerge it into the melted agarose (Figure 1D). Quickly cool the tube on ice to solidify the agarose. The embedding and cooling process should take less than 2 minutes.
- VF300 tissue slicer (Precisionary Instruments, Greenville, NC) is used to make sections. Cut the agarose block containing the VNO in shape and glue it to the tissue holder (Figure 1E and F). Insert the tissue holder into the metal cylinder and fill the remaining chamber with additional LMA. Once the LMA solidifies on ice, proceed to sectioning immediately. Supply cold oxygenated mACSF into the sectioning chamber and start cutting at 180-200 μm thickness per slice. Adjust the advancing speed and vibration frequency so that the tissue is not compressed during sectioning and VNO does not fall off from the agarose. Collect and transfer the sectioned slices to mACSF incubation chamber (Figure 2A). The slices are viable for 6-8 hours in oxygenated mACSF at room temperature. Figure 2B and C show DIC and 2-photon images of a G-CaMP2 VNO slice, respectively.
3. Imaging chamber set up
- Place the VNO slice in the middle of the perfusion chamber (Siskiyou, Grants Pass, OR) and hold the slice down with a slice anchor (Warner Instruments, Hamden, CT) (Figure 3A). The threads of the anchor should only press against the LMA part of the slice but not the VNO tissue. Oxygenated mACSF is delivered to the perfusion chamber through inlet port at ∼100 μl/sec and the liquid is drained through a suction needle via the outlet port to provide a continuous flow of fresh mACSF (Figure 3A and B).
- Fill a 30 ml syringe with Ringer’s solution and clamp it to the syringe pump (New Era Pump Systems, Farmingdale, NY). Set the pump speed to 300-600 μl/min to provide a continuous flow of Ringer’s solution over the slice (Figure 3C).
- Connect the outlet of the Ringer’s to a HPLC injection loop (Chromtech, Apple Valley, MN) (Figure 3D). Different loop size can be selected for precise control of the volume being delivered. We use a 20 μl loop in our experiments. The injection loop control has two flow routes (Figure 3E). At the “load” position, samples can be loaded into the sample loop with a Hamilton precision syringe. Excess sample exits the sample loop through waste outlet. Ringer’s solution injected by the syringe pump bypasses the sample loop and goes directly to the outlet, which is connected to the perfusion tip (Figure 3A and E). At “injection” position, the pump solution flows through the sample loop and push the stimuli into the outlet (Figure 3E). Prior to the imaging experiment, air bubbles should be chased out to ensure smooth flow of the perfusion fluid. Other commercial or custom made injection systems can also be implemented for stimulus delivery.
- Adjust the perfusion tip under a 5X or 10X lens so that the tip is about 1 mm away from the VNO slice.
- When a new perfusion system is setup, it is advisable to measure the sample delay time and duration with fluorescent dye. Load 0.1% rhodamine 6G dye into the sample loop and switch the valve to inject position at 5 seconds after the start of image acquisition (Figure 3F). The fluorescent signal is detected between 10-30 seconds. The smooth curve of fluorescent signal also indicates that there is little turbulence generated under the dipping lens and that adequate perfusion of oxygenated mACSF reaches the slice.
4. Time lapse imaging
- We use the Zeiss AxioSkope FS2 microscope with a 10X or 20X water-dipping lens for time lapse imaging. Standard GFP bandpass filter (450-490nm) is used for G-CaMP2 signals. The epifluorescent images are acquired by a CCD camera (Zeiss HRM) with 1X1 or 2×2 binning depending on the expression levels of G-CaMP2.
- Set the acquisition speed to 1 frame per second. Adjust the intensity of the light to minimize bleaching of the G-CaMP2 signals and photo damage to the cells. For each experiment, we normally acquire a 60-frame image stack (∼60-70 seconds). Switch the valve to injection position at a specific time point (eg, 5 second in Figure 3F) for one set of experiment to obtain consistent time delay in all trials.
- Perform a test run using Ringer’s solution as the stimulus. Re-adjust the perfusion setup if movement artifact is introduced during sample injection.
- Perform a positive control run with mouse urine diluted at 1:100 in Ringer’s solution. The typical maximal ΔF/F value of G-CaMP2 response to mouse urine is around 20-40%. If needed, one can confirm the viability of the slice by delivering 10 mM KCl in Ringer’s to stimulate the slices at the end of the experiments.
- Wash the Hamilton syringe in Ringer’s solution at least three times after loading one stimulus. Wash the sample loop with Ringer’s solution at least three times between different stimuli. These steps prevent cross contamination among different samples.
- Wait for 4-10 min for the VSNs to recover before applying the next stimulus.
5. Data analysis
- Perform image registration of all the images acquired from one slice. We use a custom-written VBA script in AxioVision (Carl Zeiss, North America) to automate this process. All image frames within the same experiment are registered against a common chosen reference frame with elastic registration (Figure 4A). Elastic registration, also known as nonlinear registration, is a category of image registration technique emphasizing the transformation of a target image non-rigidly to a reference image. Here we used the implementation from AxioVision.
This reference frame is chosen arbitrarily from the image stacks. - Perform image subtraction to identify the responding cells. We use custom written macros in ImageJ v1.42 (http://rsb.info.nih.gov/ij/, NIH, Bethesda, MD) to automate this process. A minimal projection image is generated for each stack. Responding cells emerge after the minimal projection is subtracted from the raw stacks (Figure 4B).
- Identify region of interest (ROI) from the subtracted stacks and obtain the ROI coordinates using Multi-Measure plugin from ImageJ. Process all stacks for one experiment and save all ROI coordinates in a ROI master list (Figure 4C).
- Use the ROI master list to measure cell responses from raw image stacks with custom-written macro and Multi-Measure plugin from ImageJ (Figure 4D). Plot response curves and heatmap in Matlab (Mathworks Inc., Natick, MA).
6. Representative Results
An example of the VNO imaging experiment using urine samples collected from four individual mice is shown in Figure 5. The slice responds to urine stimulation with diverse patterns of activation. About 80 cells are identified showing response to at least one of the urine stimuli and their response ΔF/F values are plotted in the heatmap (Figure 5A). The response traces of cells 1, 2 and 3 are plotted in Figure 5B showing the time course activation by urine stimuli. Cell 1 displays response to both female urine samples but not the male samples, whereas cell 2 shows the opposite patterns of activation and responds to both male samples only. Cell 3 is activated by both individual male and female samples. Cells 1 and 2 show characteristic response to sex specific cues in the urine 15.
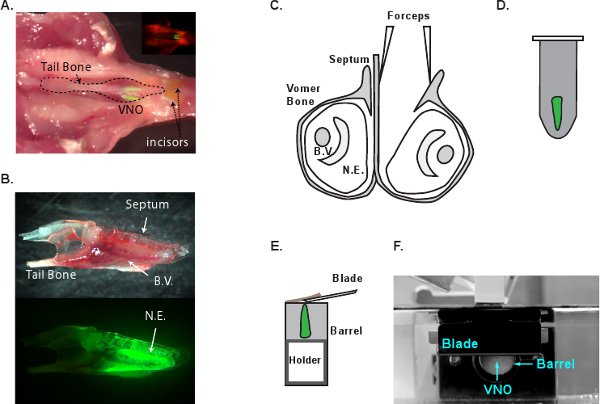
Figure 1 Schematic illustration of VNO dissection process. A. The anatomical location of VNO in the mouse head. The head of a G-CaMP2 mouse has been dissected and is laid upside down with the soft tissue removed from the palate to expose the VNO. Inlet shows a fluorescent image of the same picture. The neuroepithelia of the VNO are bright green. B. A side view of the isolated VNO that is enclosed in the vomer bone (top) and the fluorescent image of the same VNO (bottom). C. A coronal view of the VNO and dissection process. One VNO is separated from the septum and the vomer bone can then be removed to extricate the neuroepithelium. D. VNO is embedded into LMA. E. The embedded block is glued to the tissue holder for sectioning. The tissue holder is advanced at 180-200 μm per slice pushing the agarose block out of the metal barrel for sectioning. The cutting blade is positioned closely to the metal barrel. F. Side view of the VF300 tissue slicer system. B.V., blood vessel. N.E., neuroepithelium.
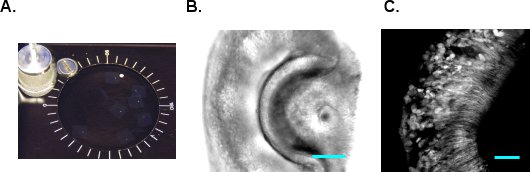
Figure 2 VNO slices. A. VNO slices are maintained in oxygenated mACSF at room temperature. B. A DIC picture of the VNO slice. C. A 2-photon image of the VNO from G-CaMP2 mouse. Scale bar, 50 μm.
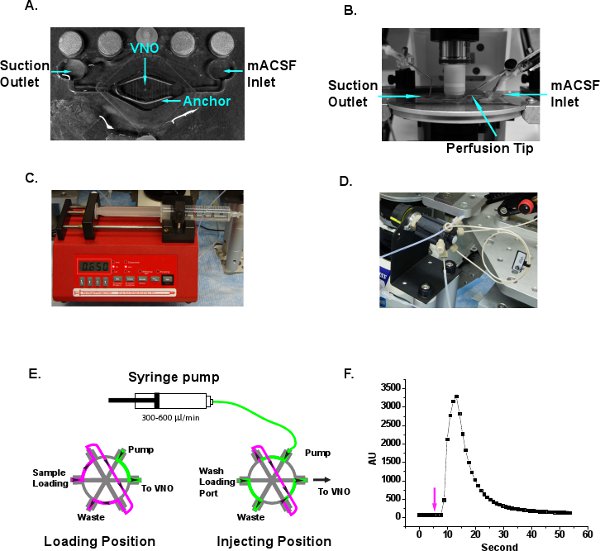
Figure 3 Illustration of perfusion system setup. A. A typical perfusion chamber with inlet and outlet. VNO slice is positioned in the center of the chamber and pressed down with a tissue anchor. The middle threads are removed from the tissue anchor so that the VNO tissue is not pressed. B. The perfusion chamber is placed on microscope stage under the dipping objective. The mACSF inlet, suction outlet and perfusion tip are indicated. C. The single barrel syringe pump provides continuous flow of Ringer’s through the perfusion tip. D. The HPLC injection loop system is adopted for convenient stimulus sample loading and injection. E. Schematic illustration of the flow directions at loading and injection positions. Ringer’s, green. Stimulus sample, magenta. Arrows indicate direction of flow. F. Delay and wash off time measurement of the perfusion system. Rhodamine 6G fluorescent dye is loaded to the sample loop and the valve is switched to the injection position at 5th second, which is indicated with an arrow. The fluorescent signal change is detected between 10-30 seconds and diminishes afterwards.
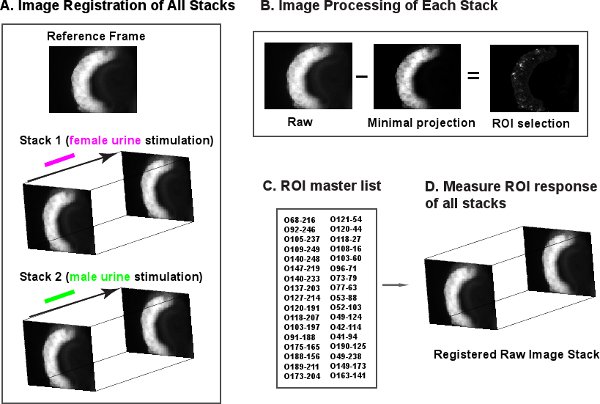
Figure 4 Image processing pipeline. A. All image frames for one slice are registered against a reference frame. B. Subtraction of minimal projection of a stack from its raw frames. Responding cells become prominent after subtraction. C. ROIs are selected and their coordinates are compiled into master list. D. Measurement of responding cells using ROI master list from raw stack.
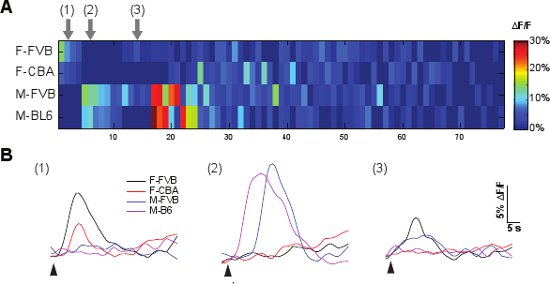
Figure 5 A representative G-CaMP2 VNO imaging experiment. A. The ΔF/F response heatmap from a slice stimulated with individual male and female urine. Urine samples are collected from individual male and female mice. X-axis, responding cells identified from the slice. Y-axis, urine samples used in the experiment. Color bar indicates the ΔF/F value. B. Response time course of 3 representative cells marked in A. Arrows indicate the time of stimulus application. F-FVB, female FVB; F-CBA, female CBA; M-FVB, male FVB; M-BL6, male C57BL/6.
Table of specific reagents and equipment:
| Name of the reagent | Company | Catalogue number | Comments (optional) |
| perfusion chamber | Siskiyou | PC-H | horizontal |
| slice hold-down | Warner Instruments | SHD-22CL | |
| perfusion port holder | ALA scientific, Inc. | MPIOH-S | |
| syringe pump | New Era Pump Systems, Inc. | NE-300 | |
| low pressure injection valve | Chromtech | V-451 | |
| PEEK tubing | Chromtech | 1531 | 1/16″ OD, 0.25mm ID |
| PEEK sample loop | Chromtech | 1803 | 20 μL |
| Hamilton syringe | Chromtech | 80630 | 100 μL |
