Caution: Refer to material safety data sheets (MSDS) before use of acetone and chloroform which are carcinogenic.
1. Preparation of Phospholipid Monolayer at a Flat Air-water Interface
- Formation of a phospholipid monolayer
- Preparation of a phospholipid solution
- Clean a 4 ml vial with a polytetrafluoroethylene coated cap using acetone, ethanol, and deionized water at least three times, and blow nitrogen gas thoroughly into the vial to get rid of water.
- Dissolve 1 mg of phospholipid (e.g., dioleoylphosphatidylcholine, DOPC) to 1 ml of chloroform in the vial to obtain 1 mg/ml of concentration. Perform this procedure in a fume hood for safety. Use lower or higher concentrations of phospholipid solution if necessary.
- Add dye tagged phospholipids (e.g., Rhodamine dipalmitoylphosphatidylethanolamine, Rhodamine DPPE) with less than 1 mol % of the phospholipid solution, in order to visualize the phospholipid monolayer with a fluorescence microscope. Perform this procedure in a fume hood for safety.
- Wrap the vial with a polytetrafluoroethylene tape to prevent solvent evaporation, and store the sample in a freezer at -20 °C.
- Deposition of phospholipids onto the air-water interface
- Clean a Petri dish (55 mm in diameter and 12 mm in height) with ethanol, and deionized water at least three times.
- Fill the Petri dish with 10 ml of deionized water to create an air-water interface.
- Spread few microliters of the phospholipid solution with a micro-syringe onto a clean interface to achieve desired surface pressure and wait for at least 30 min to evaporate the solvent completely, prior to doing the experiment.
Note: A Langmuir trough can be used instead of a Petri dish if precise control of surface pressure is necessary.
- Preparation of a phospholipid solution
- Surface pressure measurement
- Measure the surface pressure of the phospholipid monolayer with a Wilhelmy plate tensiometer. Wait for at least 30 min to get the filter paper wetted enough if filter paper is used as a Wilhelmy plate. The detailed protocol is available in Kuhn et al.20.
- Adjust the deposited amount of the phospholipid to control the surface pressure precisely. Add 4 µl of the DOPC solution (1 mg/ml) onto the 30.25 cm2–area of the interface if ~ 5 mN/m of surface pressure is required.
Note: If the filter paper is not fully wetted, surface pressure changes dramatically during the first 30 min of the experiment due to the weight change of the filter paper.
- Minimization of the convective flow of the monolayer
- Use a cone-shaped apparatus that contains a 3 mm reservoir with two thin channels, connected to a large section of the Petri dish, to suppress the convective flow of the monolayer, which can disturb morphology imaging and surface pressure measurement.
- Make sure to match the water levels between the inside and outside of the cone-shape apparatus to ensure that phospholipids move freely over the whole region before depositing phospholipids onto the air-water interface.
2. Preparation of Phospholipid Monolayer at the Curved Surface of a Droplet
- Tapering process of a glass capillary using micropipette puller
- Place a glass capillary (o.d. 1 mm, i.d. 0.78 mm, length 100 mm) on a capillary holder of a micropipette puller.
- Design a program for pulling the capillaries with appropriate parameter values (Heat: Ramp, Pull: 60, Vel: 70, Delay: 70 and Pressure 200) and pull the capillaries with the designed program according to manufacturer’s protocol.
Note: A capillary end with a few micrometers-diameter is necessary to form a 100 µm-droplet by applying a pressure with ~ 10 kPa. If the tip end of the capillary is too small, much higher pressure, > 600 kPa, is required to obtain the droplet, while it is hard to control the size of the droplets with a too large capillary tip end.
- Absorption of phospholipids onto the droplet surface
Note: To form a phospholipid monolayer at the curved interface of a droplet, a phospholipid solution without dye tagged phospholipids is used to obtain an intensity contrast, against the flat monolayer, prepared by procedure 1. In addition, both procedures 2.2.1 and 2.2.2 are allowable here. If precise control of the surface pressure at the droplet surface is necessary, procedure 2.2.2 is highly recommended. However, if not, procedure 2.2.1, which is a much easier way than procedure 2.2.2, is useful.- Coating process of phospholipids at a tip end
- Clean a slide glass using acetone, ethanol, and deionized water at least three times.
- Place the tapered capillary on a cleaned slide glass, and tilt the capillary to facilitate touching the slide glass with a tip end.
- Drop a few droplets of phospholipid solution (1 mg/ml) onto the tip end of a capillary that is adhered to the glass slide using a glass syringe, and wait at least 30 min to evaporate the solvent completely.
Note: It is highly recommended to adhere the tip end of the capillary to the glass slide during procedure 2.2.1.3 because the perimeter of the tip end is too small to hold the droplet of the phospholipids at the tip end only by capillary force.
- Preparation of a vesicle solution
- Clean a vial, and remove water from the vial, as introduced in procedure 1.1.1.1.
- Dry a 2 ml volume of phospholipid solution (1 mg/ml) by applying nitrogen gas gently, and desiccate the vial for 1 hr at RT to eliminate any remaining solvent. Perform this procedure in a fume hood for safety.
- Add 2 ml of deionized water into the vial containing dried lipids, and incubate the vial in an oven at 60 °C for 1 hr.
- Shake the vial several times, and sonicate (HF-frequency: Up to 40 kHz, Power: 370 W) for 30 min to obtain vesicles.
- Perform extrusion and freeze-thaw processes to obtain monodisperse unilamellar vesicles. The detailed protocol for preparing vesicles is described in Mayer et al.21.
Note: Surface pressure of the droplet interface is controlled precisely by adjusting the waiting time after formation of the droplet that contains monodisperse unilamellar vesicles. Using a pendant drop method22, it is necessary to measure the change of surface pressure according to time, prior to doing the experiment.
- Coating process of phospholipids at a tip end
- Formation of a droplet that contains a phospholipid monolayer at the curved surface
- Fill in the tapered capillary with 10 µl of deionized water from procedure 2.2.1. Alternatively, use 10 µl of the vesicle solution from procedure 2.2.2.
- Connect the capillary to an automated micro-injector to provide the pressure to form the droplet.
- Mount the capillary that is connected to a micro-injector to a micromanipulator to control the position of the capillary precisely.
- Prepare a bright field microscope (Microscope 1, objective lens: 10X NA 0.3) for imaging the lateral view of the capillary with a CCD camera. Microscope 1 thus enables to observe the precise position of the droplet along the z-axis and estimate the size of the droplet.
- Move the tip end to a position where the lateral view of the tip end is well visualized by Microscope 1 using a micromanipulator, and apply a variable pressure (~ 100 hPa) with the tip end of the capillary, until an appropriate size (~100 µm in diameter) of droplet is formed.
Note: It is recommended that automated micro-injector and micromanipulator be used, if fine control of the droplet size and location of the droplet is necessary. It is also possible to use manual ones.
3. Imaging Fluorescence Recovery after Droplet Merging
Note: The fundamental principles of this protocol are identical to those of the FRAP technique, except for the drop coalescence process. The detailed protocol and related theories of FRAP are available in A. Lopez et al.15 and D. Axelrod et al.16.
- Monitoring and controlling the location of the droplet
- Prepare an inverted microscope set (Microscope 2, objective lens: 10X NA 0.3, tube lens: focal length 17 cm) that enables both a fluorescence microscope with a proper filter set for Rhodamine-DPPEs (excitation at 560 nm, emission at 583 nm) and a bright field microscope. Use a CCD camera here for visualizing the top views of the droplet with a fluorescence microscope mode and a bright-field microscope mode.
- Move the droplet coated with a phospholipid to a flat air-water interface along the z-axis, but do not merge the droplet yet, using the micromanipulator. Use Microscope 1 to visualize the lateral view of the droplet.
- Locate the droplet at the center of the top view of the flat monolayer, using the micromanipulator. Use the bright-field microscope mode of Microscope 2 to visualize top view of flat monolayer.
- Merging the droplet onto the flat air-water interface
- Move the droplet further toward the flat interface until the droplet merges onto the interface, using the micromanipulator. If the merging process is done successfully, a dark region with a circular shape that is surrounded by a white background, is observed using the fluorescence mode of Microscope 2.
- Record the series of fluorescent images according to time after merging the droplet, using the fluorescence mode of Microscope 2. Use a faster frame rate than the diffusion time scale of the monolayer here. In DOPC monolayer, it takes a few minutes to diffuse into the 200 µm-dark area completely.
4. Determining the Diffusion Coefficient by Image Analysis
Note: To determine the diffusion coefficient from a series of images, customized program for image analysis is built as described below. A detailed source code of this program is available in JoVE website.
- Detection of a circular region of interest
- Detection of the center of the region of interest
- Obtain a series of fluorescent images that have been recorded during a recovery process which contains a circular dark area and a white background, and set the first image of the series as a reference image. Here, RD is the radius of the dark area in a reference image.
- Convolute the reference image with a white circle whose intensity is uniform over a whole region. Use embedded function named ‘conv2’ for convolution as shown in the source code of the customized program line 124. Here, the radius of the white circle is slightly smaller than RD.
- Find a position that indicates a minimum value in the convolution calculation, and set this position as the center of the region of interest in the reference image.
- Determining a radius of the region of interest
- Convolute the reference image with a white circle that contains a center position, determined by a procedure 4.1 and uniform intensity over a whole region. Here, RS is the radius of the white circle. Use iteration such as ‘for’ or ‘while’ with equation of a circle to make the white circle as shown in the source code of the customized program line 102 to 109.
- Convolute the reference image with another white circle which has a slightly larger radius (5%–10%), RL, than RS. Use same method with procedure 4.1.2.1 to make the circle as shown in the source code of the customized program line 113 to 120.
- Obtain the difference in value between the two convolution calculations of 4.1.2.1 and 4.1.2.2.
- Repeat the above procedures from RS = RD/2 to RS = 2RD, by increasing both the RS and RL with a pixel level. Make an iteration that contains whole source codes from procedure 4.1.2.1 to 4.1.2.3 to repeat.
- Find the radius of RS that indicates the maximum difference in value between the two convolution calculations. Use embedded function named ‘max’ to find the radius indicates the maximum difference in value as shown in the source code of the customized program line 148 to 149. This RS thus indicates the radius of the dark area in the reference image.
- Detection of the center of the region of interest
- Calculation of a fractional intensity
- Set a circle that contains a center position and a radius, determined by procedure 4.1 as a region of interest.
- Calculate the average intensities of the region of interest in a series of fluorescence images according to time. Convolute each frame with region of interest and divide it by area of region of interest to calculate the average intensity. The details are available in the source code of the customized program which is available in JoVE website.
- Calculate a fractional intensity, defined as the equation below, where F(t) is the average intensity in the circle that is a region of interest according to time, Fi is the initial intensity in the circle, and Fo is the intensity of a white background.
f(t) = (F(t) – Fi)/(Fo – Fi) (Equation 1).
- Fitting the fractional intensity to FRAP theory
- Fit the fractional intensity with the equation below, where τ is a characteristic diffusion time and I0, I1 are modified Bessel functions, using a fitting program, and obtain τ
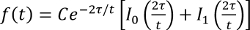 (Equation 2).
(Equation 2). - Obtain a diffusion coefficient based on the relation, τ = a2/4D, where D is the diffusion coefficient and a is the radius of the dark area.
Note: During the fitting process, it is possible to shift the fractional intensity profile along a time axis when the recovery process has already started, before recording the images.
- Fit the fractional intensity with the equation below, where τ is a characteristic diffusion time and I0, I1 are modified Bessel functions, using a fitting program, and obtain τ
A series of fluorescence images were obtained with time during the recovery process after merging a droplet coated with a DOPC monolayer onto a flat DOPC monolayer, as listed in Figure 1. The DOPC monolayer at the flat air-water interface was doped with low amounts of Rhodamine-DPPE, and this thus made it possible to visualize a background with a bright color and a dark region newly added to the flat interface. There, a recovery process was observed at 23 mN/m of fixed surface pressure. A fit of equation 1 to the change of fractional intensity according to time is shown in Figure 2. The R2 value of this fit is 0.999, and this fit still works well even at lower or higher surface pressures. The diffusion coefficient of the DOPC monolayer obtained from this fit was 27.54 µm2/sec at 23 mN/m of surface pressure.
For further validation of the FRAM, diffusion coefficients of the DOPC and dipalmitoylphosphatidylcholine (DPPC) monolayers were measured according to surface pressure. As shown in Figure 3, FRAM captures the rapid decrease of diffusion coefficient of the DPPC monolayer at ~ 9 mN/m of surface pressure, where a LC (liquid condensed) – LE (liquid expanded) phase transition occurs10, and the values of diffusion coefficients are also agreed well with the previously measurements by Peters et al.19. In addition, an exponential decay of the diffusion coefficient with the surface pressure was observed in the DOPC monolayer, and this tendency is almost identical with the one measured by Caruso et al.12.
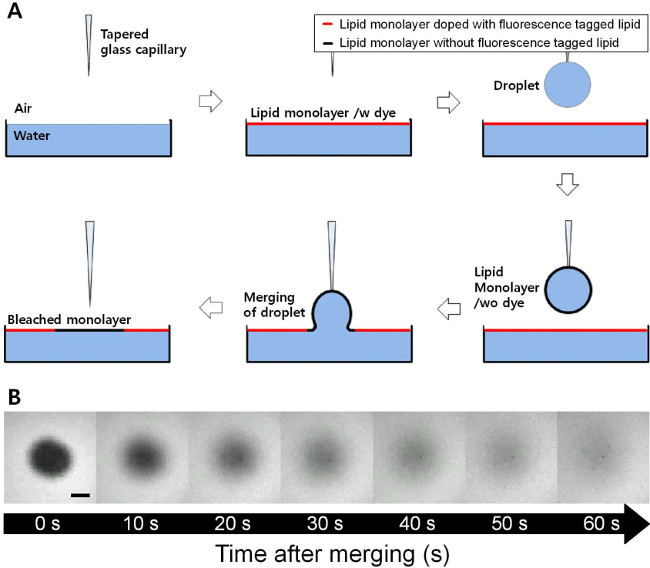
Figure 1. (A) Schematic illustration of the FRAM (fluorescence recovery after merging) technique. As the lipid monolayer coated droplet is merged to flat air-water interface, the lipid monolayer without fluorescence tagged lipid is inserted into flat fluorescent lipid monolayer. (B) Fluorescence microscope images with time during the recovery process of a DOPC monolayer at 23 mN/m of surface pressure (scale bar = 100 µm). The dark region of the DOPC monolayer is fully recovered by the diffusion process within several minutes. Please click here to view a larger version of this figure.
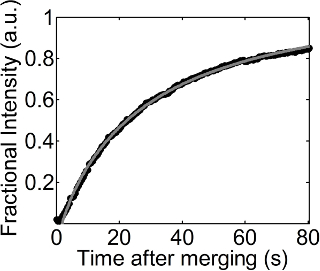
Figure 2. Fractional intensity vs. time. Black circles and the gray dotted line indicate the values of fractional intensities obtained from image analysis (procedures 4.1 and 4.2) and the fit of equation 1 to the fractional intensities with time, shown in procedure 4.3, respectively. Please click here to view a larger version of this figure.
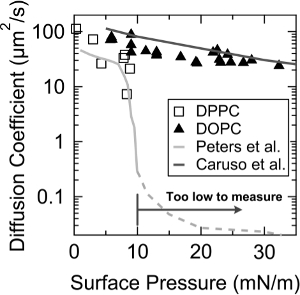
Figure 3. Diffusion coefficients of DOPC (triangle) and DPPC (empty square) monolayers as a function of surface pressure. The lines with the bright gray color and with the dark gray color indicate the values of the diffusion coefficient previously reported by Peters et al. and Caruso et al., respectively. This figure has been modified from Jeong et al.23. Reprinted with permission from Jeong et al. Langmuir. 30(48), 14369-14374. Copyright 2014 American Chemical Society. Please click here to view a larger version of this figure.
