The dual immunostaining of GAP43 (red) and GFAP (green) was used to analyze the shape of neurons and glial cells, respectively2,5. Both neurons and glial cells are present in CGN culture. The GFAP-positive glial cells are large and irregular in shape, as indicated by the arrows in the images (Figure 1). Traditional assays for cell vitality cannot distinguish glial cells from neurons when used to measure neuronal viability. Therefore, FDA-PI and FDA-PI-Hoechst staining, which can distinguish glial cells from neurons, are advantageous for the accurate evaluation of neuronal viability in CGN culture.
The low-potassium challenge was used to induce neuronal death in a CGN culture. The representative images show the CGN culture challenged with a low-potassium medium (5K), normal medium (25K), or original medium, as analyzed by FDA-PI double staining (Figure 2A) or FDA-PI-Hoechst triple staining (Figure 2B). Neuronal viability measured by various methods is presented in Figure 3 (mean ±SEM). The cell viabilities measured by FDA-PI double staining in the control, 25K, and 5K groups were 99.8 ±4.2%, 98.2 ±2.9%, and 43.9 ±8.6%, respectively (Figure 3A). The cell viabilities measured by FDA-PI-Hoechst triple staining in the control, 25K, and 5K groups were 99.8 ±1.6%, 96.7 ±4.4%, and 48.3 ±4.4%, respectively (Figure 3B). The cell viabilities measured by MTT assay in the control, 25K, and 5K groups were 102.1 ±3.9%, 96.5 ±1.7%, and 57.5 ±5.7%, respectively (Figure 3C). The percentages of lactic dehydrogenase (LDH) release in the control, 25K, and 5K groups were 100.0 ±5.5%, 94.5 ±11.2%, and 202.1 ±15.3%, respectively (Figure 3D). Neuronal viability could not be directly calculated by the LDH assay2. The MTT assay assesses the activity of NADPH-dependent cellular oxidoreductase, which reflects the number of viable cells2. However, this method could not distinguish glial cells from neurons when used to measure neuronal viability in CGN culture. By using FDA-PI and FDA-PI-Hoechst staining, the number of glial cells could be excluded, and neuronal viability could be accurately measured. Moreover, the neuronal viability in 5K medium-treated CGNs measured by FDA-PI or FDA-PI-Hoechst staining was slightly smaller than that measured by MTT assay. This might be because most glial cells that were not sensitive to 5K medium-induced neurotoxicity were excluded by FDA-PI or FDA-PI-Hoechst staining, but not by the MTT assay.
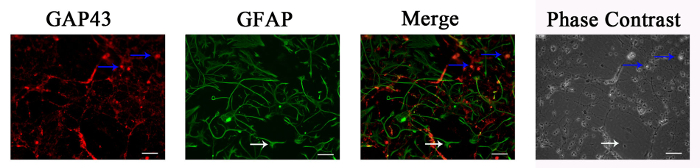
Figure 1: Both Small Neurons and Large, Irregular Glial Cells are Present in CGN Culture. At day 8 in vitro, CGNs were dual immunostained with GAP43 (red) and GFAP (green) for neurons and glial cells, respectively. The phase contrast images show the morphology of the cells. Traditional assays for cell vitality cannot distinguish glial cells from neurons when used to measure neuronal viability. Therefore, FDA-PI and FDA-PI-Hoechst staining, which can distinguish glial cells from neurons, are advantageous for the accurate evaluation of neuronal viability in CGN culture. Scale bar: 100 µm. Blue arrow: typical neurons; white arrow: typical glial cells. Please click here to view a larger version of this figure.
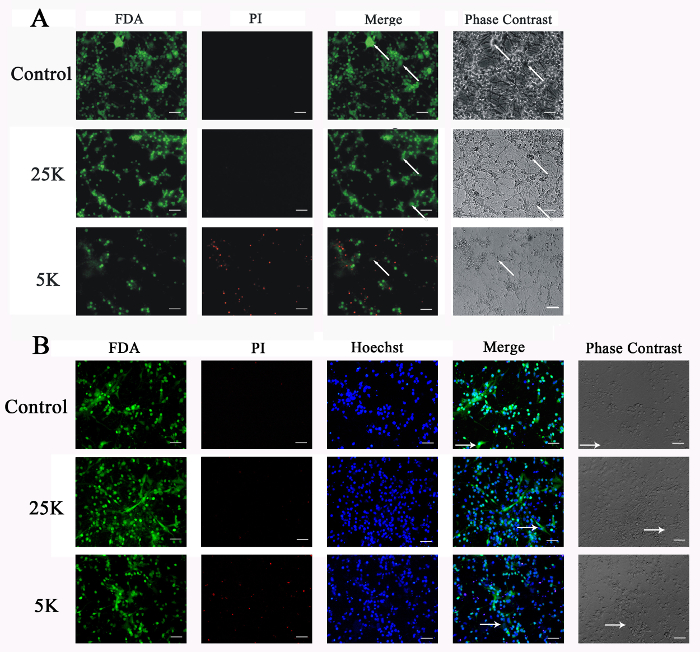
Figure 2: FDA-PI Double Staining and FDA-PI-Hoechst Triple Staining Demonstrate Low-potassium-induced Neuronal Death in CGN Culture. At day 8 in vitro, CGN cultures were switched to 5K or 25K medium. The medium in the control cultures was not changed. After 24 h of challenge, the CGN cultures were assayed by (A) FDA-PI double staining or (B) FDA-PI-Hoechst triple staining. Scale bar = 100 µm. Arrow: typical glial cells. Please click here to view a larger version of this figure.
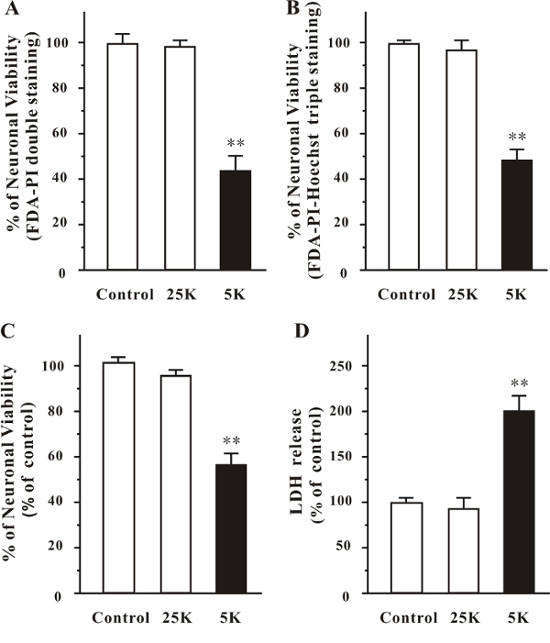
Figure 3: Quantification of Neuronal Viability in CGN Culture. At day 8 in vitro, CGN cultures were switched to 5K or 25K medium. The medium in the control cultures was not changed. After 24 h of challenge, cell viability was analyzed by (A) FDA-PI double staining, (B) FDA-PI-Hoechst triple staining, and (C) MTT assay. (D) The LDH release was analyzed by LDH assay. The data are expressed as the means ±SEM. **p <0.01 versus control (ANOVA, Tukey's test). Please click here to view a larger version of this figure.
