This protocol describes how to use the C. elegans excretory canals to visually and molecularly analyze unicellular tubulogenesis and intracellular lumen morphogenesis in a single cell. During their extension from the time of mid-embryogenesis to adulthood, the four excretory canals continue to expand their basolateral and apical/lumenal membranes together with their canalicular and endosomal endomembrane system, providing a unique model for the in vivo analysis of de novo polarized membrane biogenesis (Figure 1). Subcellular components such as apical membranes, the cytoplasm, and endosomal versus canalicular vesicles can be visualized by expressing specific fluorescent fusion proteins (described in protocol section 1), and they can be distinguished from each other by double or triple labeling in a single transgenic animal (Figure 2). A unique excretory canal phenotype (Figure 3A–B) generated by overexpressing an apical membrane associated molecule (ERM-1), is used to demonstrate how to perform a targeted RNAi screen to identify genes functioning in apical membrane and lumen biogenesis; this serves as an example of a molecular and visual analysis of polarized membrane biogenesis in this model (described in protocol section 2). This ERM-1[++] canal phenotype is used to demonstrate how to visually evaluate and score suppression (Figure 3C–D) and enhancement (Figure 3E–F) by dissecting fluorescence microscopy (described in protocol section 3). Canal phenotypes induced by modulation of ERM-1 levels serve to show how to resolve defects at the subcellular level by confocal microscopy (Figure 4) (described in protocol section 4), and how to quantify simple canal phenotypes (canal length and cyst size) and apical membrane biogenesis defects (Figure 5).
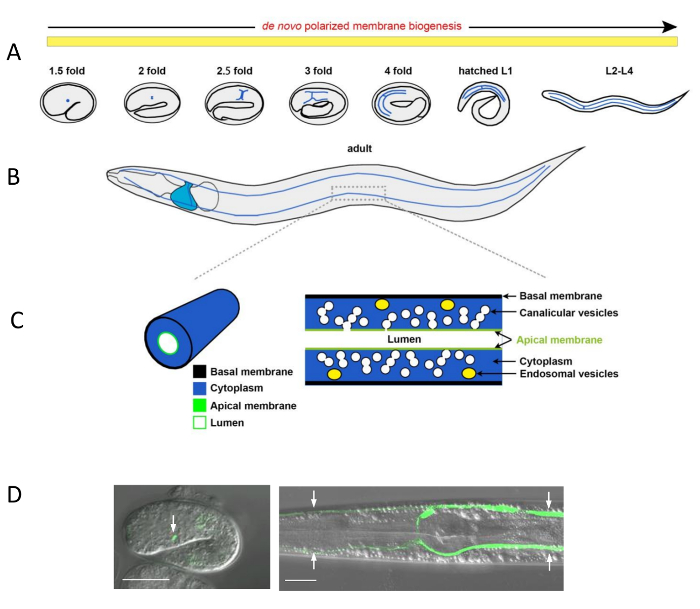
Figure 1: Morphogenesis of the C. elegans Excretory Canal and Subcellular Canal Structures
(A) Schematic representation of excretory canal extension (blue lines) during development of the embryo and larva. The excretory cell reaches its final location at the left ventro-lateral side of the posterior pharyngeal bulb at the comma stage of the embryo (not shown). As the embryo elongates, it first extends two arms laterally towards the left and right sides; then each arm bifurcates into an anterior and posterior branch. These anterior and posterior branches extend further throughout elongation of the animal during four larval stages, first catching up with the animal’s growth at the L2 stage, then accompanying its further growth, until adulthood. De novo polarized membrane biogenesis supports this extension up to adulthood. (B) In the adult animal, the anterior canal branches reach the nose tip and the posterior branches, the tail (blue lines). The excretory cell body is shown near the posterior pharyngeal bulb (blue). Polarized membrane domains are maintained during adulthood. (C) Enlarged views of a canal arm section. Left: 3D view of the canal shows: basal membrane (black), cytoplasm (blue), lumenal membrane (green) and the lumen (white). Right: inside view of the canal and its membranes: basal membrane (black), cytoplasm (blue), endosomes (yellow ovals), canalicular vesicles (white spheres, connected to each other, connected to the lumen, or isolated single vesicles), apical membrane (green) and the lumen (white). (D) Confocal/DIC overlay micrographs of the excretory cell in an embryo and the excretory canals in a larva, labeled by lumenal versus cytoplasmic GFP, respectively. Left image: excretory cell (green, arrow) in a 1.5-fold embryo, with canal lumen visualized by expressing apical ERM-1::GFP under the erm-1 promoter. Right image: four excretory canal branches (green, arrow) in L3 larvae, visualized by cytoplasmic vha-1p::GFP. Scale bars = 20 μm. Please click here to view a larger version of this figure.
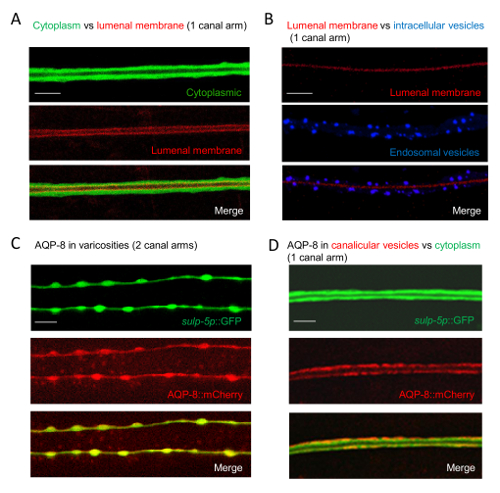
Figure 2: Visualizing Subcellular Components in Wild-type Excretory Canal Arms by Double Labeling with Fluorescent Fusion Proteins
(A) Distinguishing the cytoplasm from the apical membrane. Expression of GFP under the sulp-5 promoter visualizes the canal cytoplasm (green, top); expression of ERM-1::mCherry under its own promoter visualizes the apical membrane (red, middle); merging these images (bottom) distinguishes between cytoplasm and the apical membrane that would otherwise be indistinguishable by single labeling even at high magnification. Scale Bar = 5μm. (B) Determining the spatial relationship of endosomal vesicles to the lumen. Visualization of the lumen by an apical membrane-associated GFP fusion protein (pseudo colored to red, top); visualization of endosomal vesicles by expressing mCherry::RAB-7 (pseudo colored to blue, middle); merging these images (bottom) shows the relative spatial positions of endosomal vesicles to the lumen. Scale Bar = 5μm. (C and D) Resolving canal tube shapes versus subcellular canal components at different magnifications. The cytoplasm of L1 larvae is labeled by expressing GFP under the sulp-5 promoter and cytoplasmic canalicular vesicles by expressing the water channel AQP-8::mCherry under the aqp-8 promoter. (C) Images acquired at lower magnification resolve a “beads-on-a-string” pattern corresponding to stage-specific varicosities (varicosities are reservoirs abundant in canalicular vesicles and other components required for canal growth), but do not resolve cytoplasmic AQP-8 puncta. (D) Images acquired at higher magnification resolve AQP-8 puncta in the canal cytoplasm, corresponding to canalicular vesicles. Scale Bars: 20 μm in C and 5 μm in D. All panels show confocal projections of 10–15 sections each. Please click here to view a larger version of this figure.
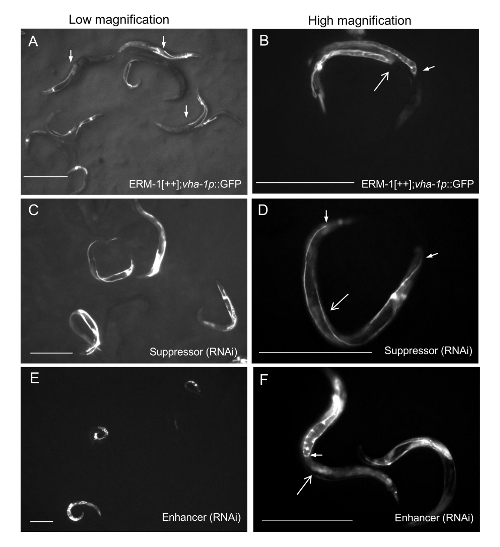
Figure 3: Scoring Enhancers and Suppressors of a Cystic Canal Phenotype (ERM-1[++]) by Dissecting Microscopy
(A) ERM-1[++]; rol-6(su1006) excretory canals are visualized by expressing GFP under a vha-1 promoter. Posterior canals extend up to the vulva or mid body of the animal (arrows; considered as 1/2 extension) at lower magnification (1.5X objective and low zoom). (B) At higher magnification, the signature ERM-1[++] small-cystic crimped canal can be resolved, with the tip of the posterior canals (one arm indicated by small arrow) extending up to the vulva or slightly beyond (vulva indicated by large arrow; canal lumen in panel D can be considered as wild type for comparison). (C) Suppression, low magnification: elongated and thin canals in RNAi treated ERM-1[++] animals. (D) At higher magnification, posterior canals can be seen to almost fully extend to the tail (small arrows; compare to that of the parent animals, shown in panel B); large arrow indicates position of the vulva. (E) Enhancement: further shortened canals with larger cysts in RNAi treated ERM-1[++] animals; low magnification, no zoom. (F) At higher magnification, posterior canal extension can be determined as less than 1/2 (small arrow) or not extended up to the vulva (indicated by large arrow) and cyst size as exceeding 1/3 of the animal’s width (wider than that of the parent animal shown in B). Scale bars= 400 µm. Please click here to view a larger version of this figure.
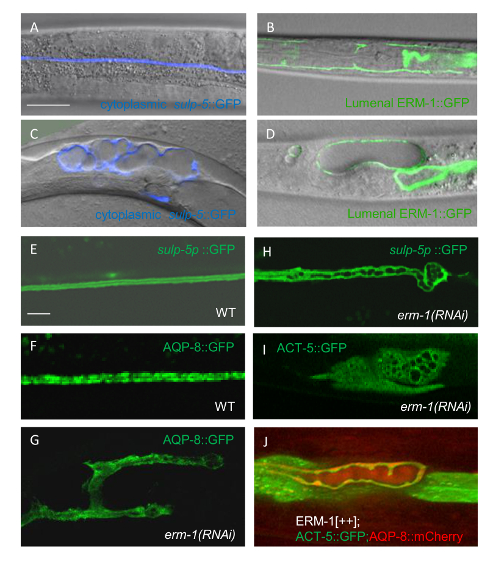
Figure 4: Visual Analysis of Excretory Canal Morphology and Subcellular Canal Components in Mutant/RNAi Animals by Confocal Microscopy
(A–D) Distinguishing a vacuolar from a cystic canal phenotype (confocal/DIC overlay micrographs are shown). (A) The cytoplasm of a wild-type canal is visualized by expressing GFP under the sulp-5 promoter (pseudo-colored to blue in distinction from apical membrane labeling, shown in green in panel B). (B) The apical membrane of a wild-type canal is visualized by expressing ERM-1::GFP; note that lumen is not visible at this magnification and that single labeling cannot distinguish cytoplasmic from membrane labeling. (C) Excretory canal phenotype induced by RNAi: cytoplasmic labeling cannot distinguish cytoplasmic vacuoles from intralumenal cysts. (D) Excretory canal phenotype induced by RNAi: Apical ERM-1::GFP labeling detects fluid accumulation inside the lumen, identifying (lumenal) cysts rather than (cytoplasmic) vacuoles (compare Figure 4I for cytoplasmic vacuoles). Scale Bar = 40 μm for A–D, shown in A. (E–J) Analyzing loss- and gain-of-function effects on subcellular canal components (confocal images of single canal arms are shown). (E-G) Effect of erm-1 RNAi on the subcellular localization of canaliculi. (E) Wild-type canal cytoplasm; note GFP exclusion indicating lumen. (F) Wild-type cytoplasmic AQP-8::GFP puncta, corresponding to canalicular vesicles. (G) Cytoplasmic and basal displacement of AQP-8::GFP puncta in erm-1(RNAi) animal. (H) Discontinuous lumen and detail of lumen tip pathology (curling and loss of lumen centering) in erm-1(mildRNAi) animal (see3 for modulating RNAi strength); note that canal cytoplasm extends beyond the tip of the lumen, here indicated by small cysts. (I) Cytoplasmic vacuoles in excretory canal body without any canal extension in strongly affected erm-1(RNAi) animal3; note that ACT-5::GFP is not recruited to the lumen (except several small specks around some vacuoles). (J) Recruitment of excess ACT-5::GFP and AQP-8::mCherry to the lumen in triple transgenic animal overexpressing ERM-1 (note thick belt of ACT-5::GFP and overlapping AQP-8::mCherry clumps around the cystic lumen). Scale Bar = 5 μm for E–J, shown in E. Please click here to view a larger version of this figure.
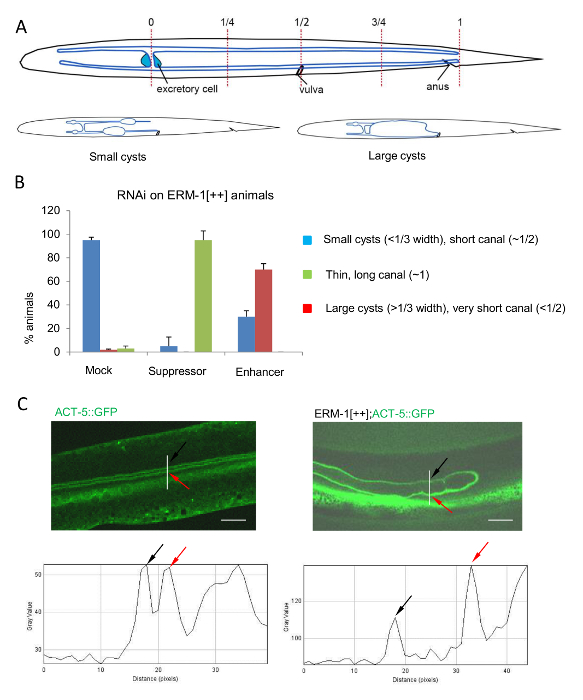
Figure 5: Examples of Quantification of Canal Defects by Dissecting and Confocal Microscopy
(A) Schematic representation of wild type (upper panel) and mutant cystic (lower panels) excretory canals for quantification of canal length and cyst size. Posterior canal length is quantified as 0, 1/4, 1/2, 3/4, and 1. Canal length ‘0’ indicates no extension and ‘1’ indicates full-length extension. Cyst size is quantified as < 1/3 (small) and > 1/3 (large) of body diameter. (B) Quantification of gross canal morphology in control and ERM-1[++](RNAi) animals by fluorescent dissecting microscopy. In mock(RNAi) ERM-1[++] animals, the posterior canal length is about 1/2 extended in 95% animals (reference image, Figure 3A–B). In suppressor ERM-1[++](RNAi) animals, the posterior canal’s length is almost fully extended in 80–90% animals (reference image, Figure 3C–D). In enhancer ERM-1[++](RNAi) animals, approximately 60-70% canals have large cysts and shorter length (< 1/2) (reference image, Figure 3E–F). Data presented as mean ± SD (n > 3). (C) Quantification of fluorescence intensity of the apical/lumenal canal cytoskeleton by ImageJ from confocal images. The canal’s apical membrane is labeled by ACT-5::GFP, wild-type canal arm is shown to the left, ERM-1[++] canal arm, to the right. Left panels: Intensity of ACT-5::GFP in wild type membrane sleeve is about 50 (gray value/membrane; indicated by black and red arrow), plot shown underneath image. Right panels: Intensity of ACT-5::GFP in ERM-1[++] is above 100 (gray value of dorsal membrane is about 110 (black arrow), and of ventral membrane is about 140 (red arrow)), plot shown beneath image. Scale bars = 5 µm. Please click here to view a larger version of this figure.
| Table 1: Examples of Markers for the C. elegans Excretory Canal Membrane System | ||||
| Protein name | Subcellular localization | Function | Examples of available strains | References |
| ERM-1 | apical domain | membrane–cytoskeleton linker | VJ402 (fgEx13[erm-1p::erm-1::gfp, rol-6p::rol-6(su1006)]) | ref (13) |
| ACT-5 | apical domain | apically localized actin | VJ268 (fgEx12[(act-5p::act-5::gfp); unc-119(+);unc-119(ed3)III]) | ref (13) |
| ABTS-2 | Basolateral | Anion/bicarbonate transporter | ABTS-2::GFP | ref (42) |
| SULP-8 | Basolateral | Sulfatepermease | SULP-8::GFP | ref (42) |
| VHA-1 | canalicular vesicles | V-ATPase | VJ535 (fgEx35(vha-1p::vha-1::gfp; rol-6p::rol-6(su1006))) | ref (9) |
| VHA-5 | canalicular vesicles | V-ATPase | ML846 (vha-5(mc38) IV;mcEx337[vha-5(+)::GFP + rol-6(su1006)] ) | ref (10) |
| AQP-8 | canalicular vesicles | aquaporin, water channel | VJ533 (fgEx33(aqp-8p::aqp-8::gfp)) | ref (9) |
| RAB-5 | endosomal vesicles | trafficking | BK209 (qpIs99 (exc9p::mCherry::rab-5)) | ref (36) |
| RAB-7 | endosomal vesicles | trafficking | BK210 (qpIs100 [Pexc-9::mCherry::rab-7]) | ref (36) |
| RAB-11 | endosomal vesicles | trafficking | BK205 (qpIs97 (exc9p::mCherry::rab-11) ) | ref (36) |
| 1Examples are selected from resources listed in Table 3. | ||||
| Table 2: Examples of C. elegans Excretory Canal-specific Promoters | ||
| promoters | Expression stage | |
| erm-1 | comma stage embryo | |
| sulp-5 | 3-fold embryo | |
| vha-1 | 2-fold embryo | |
| vha-5 | 2-fold embryo | |
| aqp-8 | 2-fold embryo | |
| glt-3 | late embryo | |
| 1Examples are selected from resources listed in Table 3. | ||
| Table 3: Resources | |||||
| Resources to identify C. elegans excretory canal-specific molecules, labeling reagents/strains and antibodies | |||||
| 1. Caenorhabditis Genetics Center (CGC)43 for available reagents and strains | |||||
| 2. Wormbase44 for information about excretory canal-specific molecules, strains and antibodies | |||||
| 3. Information about excretory canal-specific molecules: see reference6 | |||||
| 4. Transgeneome website45 for translational GFP fusion constructs | |||||
| 5. C.elegans expression pattern46 for transcriptional GFP fusion constructs | |||||
| 6. National BioResource Project (NBRP)::C.elegans47 for information on C. elegans mutants and promoters | |||||
| 7. Fluorophores: see reference48,49 | |||||
| Web-based resources for assembling molecules for a targeted RNAi library | |||||
| 1. GeneMANIA50 | |||||
| 2. AceView51 | |||||
| 3. iHOP52 | |||||
| 4. Wormbase44 | |||||
| 5. Saccharomyces Genome Database53 | |||||
| 6. Flybase54 | |||||
| 7. Mouse Genome Database55 | |||||
| 8. Human Genome Database56 | |||||
| 9. BLAST57 |
| Table 4: Example of Simple Phenotype Scoring Sheet | |||||||
| RNAi library | Strain | General Phenotype (% total) | Canal phenotype | ||||
| Canal length < 1/2 (%L4) | Canal length >1/2 (% L4) | Other Canal phenotype | Total number of animals counted | ||||
| Plate No | Well number | ||||||
| I-1 | A1 | ERM-1[++]; vha-1p::GFP | Clr (30) | 80 | 20 | 100 | |
| X-5 | B12 | same | none | 30 | 70 | 100 | |
| III-10 | C5 | same | Unc (54) | 70 | 30 | large cysts | 100 |
