All experimental procedures were conducted in accordance with the National Institutes of Health guidelines and were approved by the NICHD Animal Care and Use Committee (ACUC). The protocol described below utilizes Nkx2.1-CreC/+;Ai9+/- pups to harvest MGE-derived interneuron precursors, but can be performed on any desired fluorescent reporter mouse line. Both male and female early postnatal mice (P0-P2) were used indiscriminately for donor and host tissue.
1. Solution Preparation
- Prepare sucrose artificial cerebrospinal fluid (sACSF) according to the following recipe (unit in mM): 87 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 10 glucose, 75 sucrose saturated with 95% O2, 5% CO2 at pH=7.4.
- Fill a beaker with 350 mL pure ddH2O based on the final desired volume (500 mL of sACSF should be enough for 1-2 litters), add a magnetic stir bar and place the beaker on the stir plate.
- Weight all the salts (NaCl, NaHCO3, KCl, NaH2PO4, glucose, sucrose), one at the time and add to the water according to the following table. Amounts can be adjusted depending on the final desired volume.
Reagent Molecular Weight Concentration (mM) Grams/500 mL Sodium chloride 58.44 87 2.54 Sodium bicarbonate 84.01 26 1.09 Potassium chloride 74.55 2.5 0.09 Sodium phosphate monobasic 119.98 1.25 0.08 Glucose 180.16 10 0.9 Sucrose 342.3 75 12.84 - Bubble the solution with 95% O2, 5% CO2 (carboxygen) for at least 20-30 mins before adding the appropriate amount of CaCl2 (0.25 mL from a 1 M solution for 500 mL sACSF) and MgCl2 (3.5 mL from a 1 M solution for 500 mL sACSF).
- Check the pH using a standard pH meter. If prepared correctly, the solution should have pH = 7.4.
- Bring the solution to final volume of 500 mL with pure ddH2O in a graduated cylinder.
- sACSF can be prepared up to 1-2 days ahead. Before use, place on ice and bubble with carboxygen for at least 15 mins before start of dissection, and keep the bottle of sACSF bubbling throughout the dissection.
2. Dissection Preparation
- The following tools should be sterilized/autoclaved: at least one small fine sharp scissors, 2 fine forceps (Style #5), curved fine forceps, brain sectioning matrice mold and several razor blades. A small spatula to remove the brain from the skull is optional.
- Set up the working station near dissection microscopes with petri dishes (9 cm and 4 cm diameters), 50 mL conical plastic tubes to collect the isolated brain regions and large bore plastic transfer pipettes, all kept on ice. Prepare several 50 mL tubes full of carboxygenated sACSF on ice. If dissecting more than one brain region, make sure to properly and clearly label both the collection tubes and the transfer pipettes to avoid contamination.
- Have an additional ice bucket for ice anesthesia of pups via hypothermia, and a proper hazardous waste bag to dispose of carcasses.
- Prepare fire-polished glass Pasteur pipettes for tissue trituration. Use a bunsen burner to sterilize and shape the tip of the pipette. Prepare several pipettes with different bore openings: large (~600 µm), medium (~300 µm) and small (~100 µm). Test out flow through tips using water to ensure different trituration speeds. At least one pipette of each kind is needed for each isolated brain region, but having several extra for each size is recommended in case of clogging or breaking tips.
3. Transplantation Preparation
- Use an electrode puller to prepare fine-tipped glass micropipettes. Break or bevel the tip to make a sharp angled point, with the tip having a diameter of ~20-40 µm. Visually inspect the tips with a microscope to make sure no dust or residual glass particles are obstructing the aperture.
- Using a fine needle syringe, completely fill the glass micropipette with mineral oil. Insert the micropipette into the Nanoject apparatus (or other microinjection device). For the Nanoject III, set up the following program: Volume = 60 nL, Rate = 30, Cycles = 25, Delay = 1 s.
- Secure the Nanoject to the manipulator that is attached to a magnetic base, and adjust the manipulator so that the Nanoject is pointing straight down, not tilted along the x or y axis.
- Attach some adhesive putty (~1-2 cm for P0-P2 pups) to the top of the cover of a petri dish creating a elevated surface to rest the pup's head on.
- Cut ~6-8 cm long stripes of lab tape and make a diamond-shaped hole in the middle.The tape will be used to stabilize the head of the pup during the procedure, while also stretching the skin and allowing easy visualization of landmarks and the targeted region.
- Prepare a heating pad next to the injection station,and turn on to 'low' setting before starting injections. Cover the pad it with some paper towels or a diaper pad.
- If performing multiple transplantation conditions, have a small scissors ready to perform toe/tail clipping to tag pups receiving different injections.
4. Removal of P0-P2 mouse brain
- Right before starting the procedure, fill several petri dishes with chilled, carboxygenated sACSF. Also fill the collection tube(s) with ~25 mL sACSF.
- Wrap a P0-P2 pup in some parafilm or a glove and place it well covered under the ice. Set a timer for 5 minutes and start it. After that time, remove the pup and check that it does not respond to pinching of the hindpaw.
- Decapitate the pup and collect the head in a petri dish filled with sACSF. Cut the skin along the midline to expose the skull.
- Locate the opening in the skull at the hindbrain/spinal cord and insert one end of the forceps along dorsal portion of the skull. Gently grasp the skull at the midline and 'peel' away pieces laterally to expose the brain, being careful not to damage the brain.
- Upon removal of the dorsal and lateral portions of the skull, use the forceps or the spatula to gently remove the brain from the skull by scooping underneath the ventral surface of the brain, trying not to damage any area. Place the brain into another petri dish filled with carboxygenated sACSF.
NOTE: We present two different strategies for dissecting out the hippocampus, striatum and cortex. The first strategy (steps 5.1-5.10) is useful for any mouse line whereas the second strategy (steps 6.1-6.7) would need a fluorescent reporter mouse to cleanly separate the striatum from the globus pallidus and other brain structures. If striatum is not desired, then ignore the striatum-specific portions of the two techniques
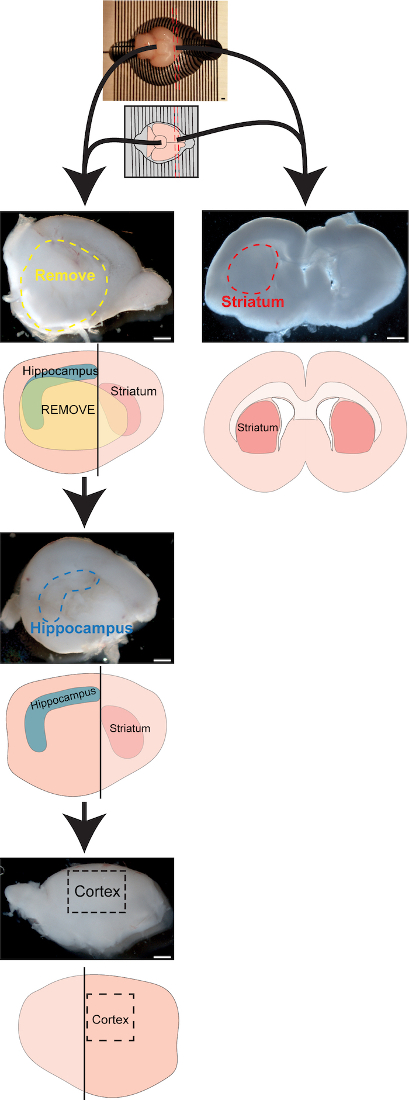
Figure 1: Schematic and images for brain dissection, technique #1
Dissection technique described in Steps 5.1-5.10. If striatal tissue is desired, place P1 brain in brain matrices ventral side up. Place razor blades into matrice slots through anterior brain to obtain coronal sections through striatum. Remove striatal pieces from both hemispheres, repeat for all sections containing striatum that are anterior to hippocampus. If striatal tissue is not desired, simply hemisect the brain, place the hemispheres medial side up in the dish, and remove the ventral-medial brain structures (thalamus, basal ganglia, etc.) to expose the hippocampus. Use tweezers to pinch of hippocampus, then flip hemisphere over and use tweezers to dissect out a chunk of cortical tissue. The vertical black lines through the schematic hemispheres where the cuts would be to remove the striatal sections. Scale bar = 500μm. Please click here to view a larger version of this figure.
5. Harvest Striatum, Hippocampus and Cortex, Technique #1
- Place whole brain on sectioning matrice mold, ventral side up. Place 1 razor blade through the most anterior portion of the brain (through anterior olfactory tract).
- Insert another razor blade just posterior to the first one to generate a 0.5 mm slice, and place an additional razor blade posterior to this one.
- Transfer these slices to a petri dish with carboxygenated sACSF and place the rest of the brain to a separate dish with sACSF. Transfer the striatal slices to a dissecting scope to visualize striatum. These slices should contain large portions of the anterior striatum and lack hippocampus. Use a fluorescent dissecting scope with Nkx2.1-Cre+/-;Ai9+/- slices to differentiate the weak striatal tdTomato signal from the strong tdTomato signal of the globus pallidus.
- With or without fluorescence, pinch out the striatum from both hemispheres on all the sections and transfer striatal chunks to properly labeled 50 mL tube with sACSF, store on ice.
- Hemisect the brain along the midline using forceps or a razor blade, and lay the hemisphere so that the medial surface is facing up.
- Remove the ventral brain tissue (thalamus, basal ganglia, etc) by pinching off this tissue with forceps. When complete, the hippocampus (a sausage shaped structure spanning the anteroposterior axis along the dorsal cortex) and ventricular side of the cortex should be clearly visible.
- To remove the hippocampus, insert the tips of the forceps in front of the anterior hippocampus and gently separate the hippocampus from the cortex by moving the forceps posteriorly and pinching along the hippocampal-cortical border. Transfer the isolated hippocampus to properly labeled 50 mL tube with sACSF, store on ice.
- To dissect the cortex, place the hemisected cortex medial side down. Then use the forceps to pinch the most dorsal, ventral, anterior and posterior portions of the cortex to leave a square chunk of medial somatosensory cortex, ~2 mm x 2 mm. Flip the cortex so medial side is up and clean of any excess non-cortical tissue (thalamus, basal ganglia, etc.) on the medial surface if necessary.Transfer the cortex chunk to properly labeled 50 mL tube with sACSF, store on ice.
- Repeat the procedure with the other hemisphere to collect both hippocampi and cortex regions.
- Repeat this procedure for all pups, making sure to replace the sACSF in the petri dishes between pups to ensure that the sACSF remains cold and fresh.
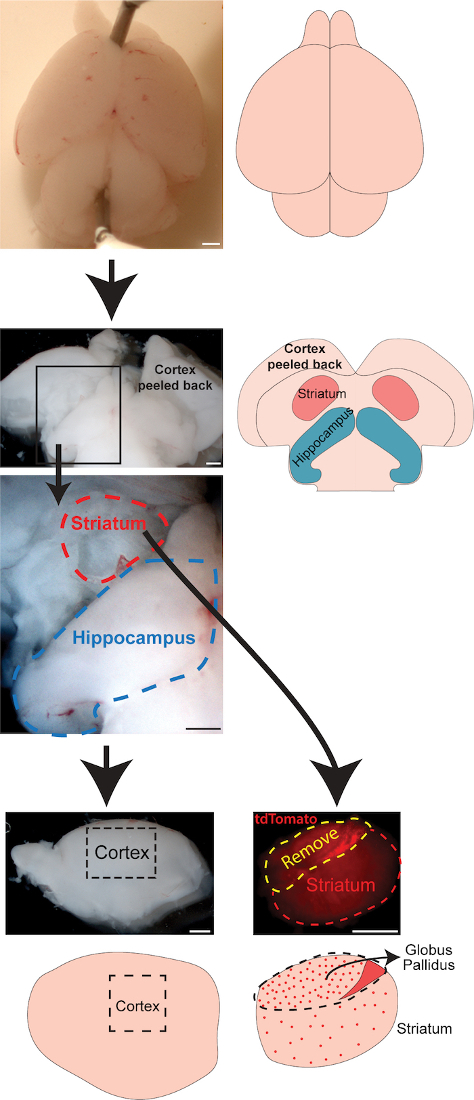
Figure 2: Schematic and images for brain dissection, technique #2
Dissection technique described in Steps 6.1-6.7. Pin brain onto a dissecting dish dorsal side up. After peeling the cortex forward, the hippocampus and striatum are visible and can be removed, then a section of cortex can be removed as described in previous technique. Depending on transgenic mouse line, striatum can be cleaned up to remove globus pallidus and other tissue. In Nkx2.1Cre;Ai9 mouse line, the globus pallidus has a significantly higher density of tomato+ cells compared to the striatum. Scale bar = 500 μm. Please click here to view a larger version of this figure.
6. Harvest Striatum, Hippocampus and Cortex, Technique #2
- Place the brain ventral side down in a sylgard-coated petri dish containing sACSF and pin it down through the cerebellum and anterior cortex or olfactory bulbs.
- Starting with one hemisphere, use curved forceps to gently separate the posterior cortex from underlying hippocampus and other tissue. Gently lay the peeled cortex flat on petri dish. Repeat for other hemisphere. Hippocampi and striatum should be clearly visible in exposed brain.
- Use forceps to pinch one hippocampus at the midline and peel hippocampus laterally to remove. Place in collection vial on ice and repeat with other hippocampus.
- For striatum, gently scrape around the edges of the striatum to loosen it from the surrounding tissue. Then remove the striatum by pinching it off from underneath and add to collection tube on ice. Repeat for other striatum.
- Cortical sections can be harvested as described in step 5.8.
- If using a transgenic reporter mouse line (e.g., Nkx2.1-Cre;Ai9, Lhx6-GFP, etc.), transfer striatum to a petri dish with sACSF and view under fluorescent dissecting scope. Use forceps to remove any non-striatal tissue from striatum (in Nkx2.1-Cre;Ai9 mice, remove all 'bright' tissue, as globus pallidus has much higher MGE-derived, tdTomato+ cell density compared to striatum). Repeat for all striatum.
- Repeat this procedure for all pups, making sure to replace the sACSF in the petri dishes between pups to ensure that the sACSF remains cold and fresh.
7. Generating Single Cell Dissociations
- After the dissection is complete, prepare a 1 mg/mL Pronase solution by weighing 10 mg Pronase and dissolving in 10 mL carboxygenated sACSF.
- Transfer the tissue from the 50 mL collection vials to 5 mL round bottom tubes containing 2 mL of the Pronase-sACSF solution. Incubate the tissue for 20 min at room temperature, shaking/flicking the tube 3-4 times with your finger during incubation to mix the samples.
- During this incubation, prepare Reconstitution solution: 1% FBS (100 μL) + DNAse (1 μL of 1:10,000 stock DNAse) in 10 mL carboxygenated sACSF.
- After the 20 mins, carefully remove the pronase solution to not disturb the tissue at the bottom, and replace it with 1-2 mL of the Reconstitution solution (total volume is dependent on starting amount of tissue).
- Mechanically dissociate the tissue by triturating the tissue with the previously prepared fire-polished Pasteur pipettes. Starting with the large bore (~600 µm) pipette, aspirate and expel the tissue solution at least ten times to break up the tissue. Do not introduce bubbles into the solution. Repeat this process for the medium bore pipette and the small bore pipette. The solution should be cloudy and no clear piece of tissue should be visible in the collection tubes.
- Pipet the cell lysate solution through a 50 µm filter into 5 mL conical tubes to remove any cell clumps. Proceed with these single cell solutions to flow cytometry (or potentially directly to cell counting if no sorting is needed).
8. Preparing FACS-purified Cell Solutions for Transplantation
- Upon receiving the cell solutions from the flow cytometer, transfer the solution to one (or multiple) 1.5 mL conical tubes and spin the cells at 500 g for 5 min at 4oC. After centrifugation, remove media so that ~20-40 µL remain in the tube. Reconstitute cells in this remaining media (and combine solution if multiple tubes were needed).
- On a small piece of parafilm, mix together 2 µL of cell solution + 8 µL sACSF + 10 µL of trypan blue stain. Pipet 10 µL into a hemocytometer with slide cover.
- Count the number of live cells in each of the 4 x 4 square grids in the corners of the hemocytometer using a standard laboratory hand tally counter (dead cells will be blue from the dye), and average these 4 counts. Multiply this number by 100 to determine the number of cells per µL.
- If needed, adjust volume so the cells are at a concentration of 10,000-30,000 cells/µL in Reconstitution solution. Final concentration is dependent on total amount of cells and desired number of transplantations. Keep cells on ice for transplantation
9. Transplantation Into P0-2 WT Pups
- Wrap a WT pup in some parafilm or a glove and place it well covered under the ice. Set a timer for 5 min and start it. After that time, remove the pup and check that it does not respond to pinching of the hindpaw.
- While the pup is on ice, mix the cell solution by pipetting it several times to ensure the cell suspension is well mixed prior to loading (mix the cells prior to loading the micropipette for every injection). Then empty the micropipette of the mineral oil and fill it completely with the cell suspension.
- Place the anesthetized pup on the petri dish with his head lying on the adhesive putty so that the top of the head is relatively flat. Place the tape with the diamond hole across the head of the pup, pulling it taut to ensure the skin is stretched and the head is firm but that the mouse is comfortable and breathing well. Lambda should be visible through the diamond hole.
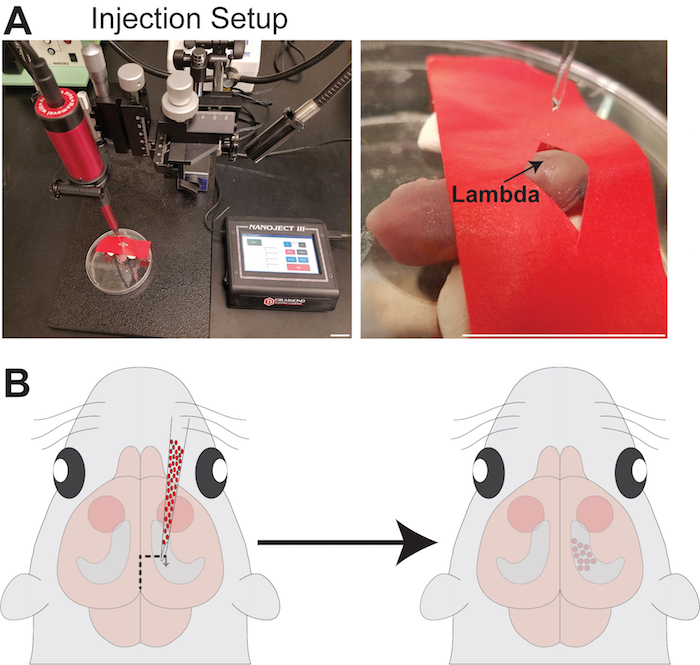
Figure 3: Schematic and images for transplantation
(A) Pictures of injection setup. Note that lambda is clearly visible through the pup's skull and should be used for zeroing the micropipette. Scale bar = 1 inch. (B) Schematic depicting the injection procedure to target the hippocampus. Please click here to view a larger version of this figure.
- Move the secured pup under the Nanoject and lower the micropipette so that the tip of the micropipette is on directly above lambda. Record the x-y coordinates on the manipulator.
- Adjust the manipulator knobs to move the micropipette to the desired coordinates along the mediolateral and the anteroposterior axis of the head: S1 Cortex → 1.0 mm anterior and 1.0 mm lateral, Hippocampus → 0.75-1.0 mm anterior and 1.0-1.2 mm lateral, Striatum → 2.0-2.2 mm anterior and 1.5 mm lateral. Coordinates can be adjusted slightly to compensate for age of the pup (e.g., P0 vs. P2) or the strain of mice (e.g., CD1 mice are larger than and C57/B6 at P2).
- Lower the micropipette until it forms a small concavity on the skin. Then turn the z-axis manipulator knob firmly but gently to drive the micropipette through the skin and skull to enter the brain. When the micropipette enters the brain, the pressure on the skull will be released and the concavity will disappear.
- Retract the micropipette slightly until the tip is surrounded by a cone of skin, shaped like a tent. Read the coordinates along the z axes: This will be the z-axis zero point.
- Lower the micropipette to the desired depth: Cortex → 0.75-1.0 mm, Hippocampus → 1.2-1.5 mm, Striatum → 2.0-2.5 mm. Depth can be adjusted slightly to compensate for age or strain of the pup.
- Inject the cell suspension using the Nanoject III program described above. After injection program is complete, wait 15-20 s before slowly retracting the micropipette to minimize solution leaking out of the injection site.
- If performing bilateral injections, re-zero the micropipette above lambda and move to the proper coordinates in the other hemisphere. Alternatively, one can make two injections into the cortex of the same hemisphere by moving the micropipette 1 mm anterior of the first injection.
- Once the transplantation is complete, remove the tape and transfer the pup onto the heating pad. If necessary, tag the pup by tail or toe clipping. Once the pup has reacquired a red color and is moving, place it back in the cage with the mother.
This protocol demonstrates how to harvest specific brain regions from early postnatal brains (Figure 1-2), collect single cell dissociations of interneuron precursors, and transplant these cells into various brain regions in naive WT postnatal pups (Figure 3). For posthoc analysis, brains that received interneuron precursor grafts were harvested between P30-35 to characterize cell morphology, neurochemical markers and electrophysiological properties. These types of assays are often carried out between P21-P30 in normal mice, but since the maturation of transplanted cells might be slightly delayed due to the dissection/dissociation procedure, waiting an additional 5-10 days is recommended to compensate for this delayed maturation. The type of analysis to be performed will dictate the proper strategy to harvest the brain. Notably, we did not observe preferential cell death of specific interneuron subgroups that could bias for or against certain subtypes20.
For immunohistochemical analysis, mice were perfused with 4% paraformaldehyde and the brains were removed. 50 μm vibratome slices were prepared through the targeted brain region and stored in antifreeze solution and/or processed for immunostaining as previously described20. Some brains did not contain any tomato+ cells, which could be due to improper targeting (e.g., injection too deep into the ventricle), cells lost or undergoing apoptosis during the grafting procedure, or rejection of transplanted cells by the host. Based on final cell counts, it is estimated that only 2-5% of grafted cells survive20, which is in line with other transplantation procedures22,23.
Not surprisingly, there was significant variability in the total number of tomato+ cells in successful transplants, ranging from dozens to several thousand tomato+ cells (Figure 4A). Grafted cells were localized in the correct regions, with many displaying interneuron morphologies and well-characterized interneuron neurochemical markers (Figure 4B). Similar cell survival numbers and maturation profiles were observed even when cells were grafted into new environments in heterotopic transplantations (Figure 4C).
In addition to immunohistochemical analysis, electrophysiological analysis on grafted cells was performed to confirm that they have integrated into brain circuitry and display expected intrinsic and firing properties. Brains were harvested from P30-35 mice and slices prepared for physiological recordings as previously described20. The grafted interneurons presented adult-like physiological properties and distinct firing patterns could be characterized that were representative of well-characterized interneuron subtypes (Figure 5A), suggesting that grafted interneurons were able to properly mature in the host environment. To verify that transplanted cells were integrated in the neuronal network, sEPSCs were also recorded (Figure 5B). In addition, a subset of transplants were performed with interneurons expressing ChR2 followed by recording from pyramidal cells localized near transplanted interneurons. These data demonstrated that postsynaptic GABAergic currents are evoked by blue light (Figure 5C-D).
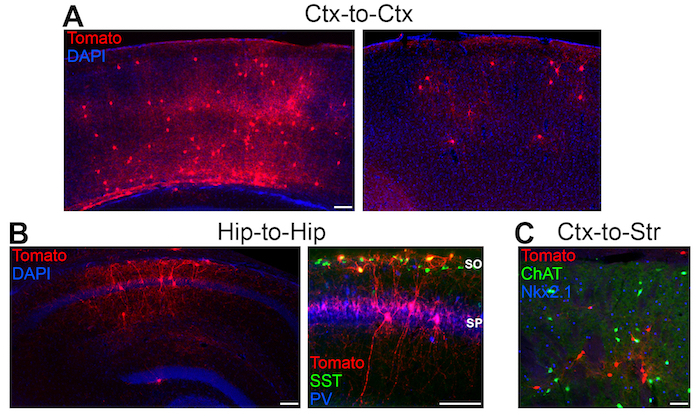
Figure 4: Grafted interneuron precursors populate host brain regions Representative sections from P30 WT mice that were transplanted with tomato+ interneuron precursors at P1. (A) In homotopic cortex-to-cortex transplantations, the grafted cells populate all cortical layers and display morphologies that mimic endogenous interneurons. Selected images highlight the variability in cell numbers from different transplants, with the left image having a much greater number of tomato+ cells per section compared to the transplant on the right side. (B) Low magnification (left) and high magnification (right) representative sections from homotopic hippocampus-to-hippocampus grafts. Note that the majority of tomato+ cells in the stratum oriens (SO) express SST (likely O-LM cells) whereas many tomato+ cells in the stratum pyramidale (SP) express PV (likely basket cells), similar to endogenous hippocampal interneurons. (C) Example of a heterotopic transplantation (Cortex-to-Striatum) with tomato+ present in the striatum. Scale bars = 200 μm in A in low mag panel in B, 50 μm in C and high power mag in B. Please click here to view a larger version of this figure.
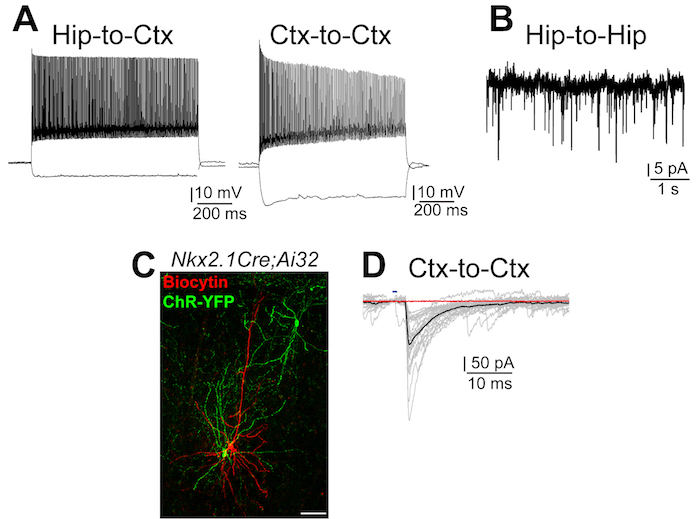
Figure 5: Grafted interneurons are electrophysiologically mature and integrate in the host neuronal network
(A) Representative examples of the highest firing frequencies recorded from grafted interneurons. Left, Fast Spiking interneuron from a Hip-to-Ctx graft, injected current steps: -100 pA and 520 pA; right, Non-Fast Spiking interneuron from a Ctx-to-Ctx graft, injected current steps: -100 pA and 360 pA. (B) Example of sEPSCs recorded in a Late Spiking interneuron from a Hip-to-Hip transplant. (C) Representative image displaying Nkx2.1-Cre;Ai32 cells (YFP) from a Ctx-to-Ctx transplant with a biocytin-filled (red) pyramidal cell. Scale bar = 50 μm. (D) Example of a GABAergic postsynaptic currents evoked by blue light pulses recorded in pyramidal cells, recorded with a [145 mM] Cl-. In black, average traces; in red, average response recorded in the presence of Gabazine. Please click here to view a larger version of this figure.
