Ethical approval was granted by the Joint Chinese University of Hong Kong―New Territories East Cluster Clinical Research Ethics Committee (Reference Number: 2010.432). Research license was approved by the Council on Human Reproductive Technology of Hong Kong (Number R3004).
1. Whole genome amplification
- Prior to start, check the volume of magnetic beads (Table of Materials) to ensure that there is no less than 135 µL (with a 20% excess) for each sample. Keep the magnetic beads at room temperature (RT) for at least 30 min. Prepare 720 µL (with a 20% excess) of 70% ethanol for each sample. Equip a thermal cycler (Table of Materials) with heated lid at 105 °C.
NOTE: The newly prepared 70% ethanol (Table of Materials) should be used up within 3 days. - Sample preparation
NOTE: In the routine practice, 5 to 10 trophectoderm cells of the blastocyst are biopsied according to the practice guideline19.- Suspend biopsies in 2 µL of 1x phosphate-buffered saline (PBS) in a single 0.2 mL polymerase chain reaction (PCR) tube.
- Briefly spin the tube on a mini centrifuge for 3 s to collect the droplets.
- Cell lysis and extraction
- Thaw the cell extraction buffer (Table of Materials) and the extraction enzyme dilution buffer (Table of Materials) on ice, vortex and briefly spin on a mini centrifuge for 3 s before use.
- Add 3 µL of cell extraction buffer to each tube from step 1.2.2.
- Prepare a 5 µL cell lysis master mix for each sample by adding 4.8 µL of extraction enzyme dilution buffer and 0.2 µL cell extraction enzyme (Table of Materials). Mix well and aliquot into each tube from step 1.3.2. Flip the tube gently and briefly spin on a mini centrifuge for 3 s.
NOTE: tips should not touch the liquid containing the cell samples when adding the master mix. - Incubate the tube from step 1.3.3 in the thermal cycler with heated lid. Run the program with the following settings: 10 min at 75 °C, 4 min at 95 °C, hold at 4 °C.
- Preamplification
- Thaw the preamplification buffer (Table of Materials) on ice, vortex and briefly spin on a mini centrifuge for 3 s before use.
- Prepare a 5 µL preamplification master mix for each sample by adding 4.8 µL of preamplification buffer and 0.2 µL of preamplification enzyme (Table of Materials). Mix well and aliquot into each tube from step 1.3.4. Flip the tube gently and briefly spin on a mini centrifuge for 3 s.
NOTE: Tips should not touch the liquid containing the DNA samples when adding the master mix. - Incubate the tube in the thermal cycler with heated lid. Run the program with the flowing settings: 2 min at 95 °C; 12 cycles for 15 s at 95 °C, 50 s at 15 °C, 40 s at 25 °C, 30 s at 35 °C, 40 s at 65 °C, 40 s at 75 °C; hold at 4 °C.
- Amplification
- Thaw the amplification buffer (Table of Materials) on ice, vortex and briefly spin on a mini centrifuge for 3 s before use.
- Prepare a 60 µL amplification master mix for each sample by adding 25 µL of amplification buffer, 0.8 µL of amplification enzyme (Table of Materials), and 34.2 µL of nuclease-free water for WGA (Table of Materials). Mix well and aliquot into each tube from step 1.4.3. Flip the tube gently and briefly spin on a mini centrifuge for 3 s.
- Incubate the tube in the thermal cycler with heated lid. Run the program with the following settings: 2 min at 95 °C; 14 cycles for 15 s at 95 °C, 1 min at 65 °C, 1 min at 75 °C; hold at 4 °C.
- Purification of the WGA products
- Transfer each WGA product from step 1.5.3 into new 1.5 mL tubes. Add 112.5 µL of magnetic beads into each tube. Vortex and incubate at RT for 5 min.
NOTE: Completely mix the magnetic beads before use. - Place the tubes onto a magnetic stand (Table of Materials) for 3 min until the supernatant is clear. Discard all the supernatant without disturbing the beads.
- Add 300 µL of 70% ethanol to each tube. Rotate each tube 180° to let the beads run through the ethanol and rotate back to the original position. Discard all the supernatant after the beads have settled without disturbing the beads. Repeat this step once.
NOTE: Keep the tube on the magnetic stand while rotating the tube horizontally. - Briefly spin each tube on a mini centrifuge for 3 s. Place the tubes onto the magnetic stand until the residual supernatant is clear. Discard all the residual supernatant without disturbing the beads. Air dry the beads at RT for approximately 3 min.
- Remove the tubes from the magnetic stand and resuspend the dried beads by adding 35 µL of low Tris-EDTA (TE) buffer (Table of Materials). Incubate at RT for 5 min.
- Place the tubes onto the magnetic stand for 3 min until the supernatant is clear. Transfer all the supernatant containing eluted DNA to new 1.5 mL tubes without disturbing the beads.
- Transfer each WGA product from step 1.5.3 into new 1.5 mL tubes. Add 112.5 µL of magnetic beads into each tube. Vortex and incubate at RT for 5 min.
2. Quality control of the WGA products
- Quantify each purified WGA product from step 1.6.6 by a fluorometer assay (Table of Materials) according to the manufacturer’s manual using 1 µL of WGA product as starting material.
NOTE: The accepted concentration of the WGA product is ≥ 10 ng/µL. Any product below this threshold is not recommended to proceed to the next steps.
3. Fragmentation of WGA products
- Prior to start, preheat a dry block heater to 37 °C. Prepare 6 µL (with a 20% excess) of 0.5 M EDTA for each sample. Based on the concentration, aliquot 300 ng of DNA from each purified WGA product in step 1.6.6 to new 0.2 mL PCR tubes and bring volume to 16 µL with nuclease-free water for each tube.
- Fragmentation
- Prepare a 4 µL double stranded DNA (dsDNA) fragmentation reaction mix for each sample by adding 2 µL of dsDNA fragmentation reaction buffer (Table of Materials) and 2 µL of dsDNA fragmentation enzymes (Table of Materials). Mix well and aliquot into each tube from step 3.1. Vortex and briefly spin on a mini centrifuge for 3 s. Incubate the tubes for 25 min at 37 °C in a thermal cycler with heated lid.
- Add 5 µL of 0.5 M EDTA immediately to each tube. Mix well by vortexing and briefly spin on a mini centrifuge for 3 s.
- Purification and resuspension
- Transfer each product from step 3.2.2 into new 1.5 mL tubes. Add 37.5 µL of magnetic beads to each tube. Mix by vertexing and incubate at RT for 5 min.
- Purify the products as described from step 1.6.2 to step 1.6.4.
- Elute each purified product as described in steps 1.6.5 and 1.6.6 by adding 32 µL of low-TE buffer.
4. Library construction
- Blunt-end repairment, size selection and purification
- Prepare a 20 µL blunt-end repairment mix for each sample by adding 9.5 µL of nuclease-free water, 10 µL of 5x end repair buffer (Table of Materialstube from step 3.3.3. Vortex and briefly spin on a mini centrifuge for 3 s.
- Add 50 µL of magnetic beads to each tube from step 4.1.1. Vortex and incubate at RT for 5 min.
NOTE: Completely mix the magnetic beads before use. - Place each tube onto the magnetic stand for 3 min until the supernatant is clear. Transfer all the supernatant to new 1.5 mL tubes where 25 µL magnetic beads are added for each. Vortex the tubes with the transferred supernatant and incubate at RT for 5 min.
- Purify the products in the incubated tubes as described from step 1.6.2 to step 1.6.4.
- Elute each purified product as described in steps 1.6.5 and 1.6.6 by adding 32 µL of low-TE buffer.
NOTE: This is a safe stop point; the purified DNA from this step is stable at 4 °C for no more than 24 h.
- Adaptor ligation and purification
- Prepare a 17 µL adapter ligation mix for each sample by adding 10 µL of nuclease-free water, 5 µL of 10x ligase buffer (Table of Materials), 1 µL of P1 adapter (Table of Materials), and 1 µL of DNA ligase (Table of Materials). Mix well by vortexing for 5 s and spin on a mini centrifuge for 15 s, and aliquot into each tube from step 4.1.5.
- Add 1 µL of adapters (Table of Materials) to each tube from step 4.2.1 according to the sample sheet (Supplemental File: Sample sheet for adapter ligation). Vortex and briefly spin on a mini centrifuge for 3 s. Incubate the tubes at RT (20−25 °C) for 20 min.
- Add 75 µL of magnetic beads to each tube from step 4.2.2. Mix by vertexing and incubate at RT for 5 min. Then, purify the products as described from step 1.6.2 to step 1.6.4.
- Elute each purified product as described in steps 1.6.5 and 1.6.6 by adding 15 µL of low-TE buffer. Transfer all the supernatant containing eluted DNA to new 0.2 mL 8-tube strips.
NOTE: This is a safe stop point; the purified DNA from this step is stable at 4 °C for no more than 24 h.
- Amplification and purification
- Prepare a 50 µL amplification master mix for each sample by adding 47.5 µL of super mix (Table of Materials) and 2.5 µL of primer mix (Table of Materials). Mix well by vortexing and briefly spin on a mini centrifuge, and aliquot into the 0.2 mL 8-tube strips from step 4.2.4.
- Vortex the strips for 30 s and briefly spin on a mini centrifuge for 3 s. Incubate the strips in the thermal cycler with heated lid. Run the program with the following settings: 20 min at 72 °C; 5 min at 95 °C; 10 cycles for 15 s at 95 °C, 15 s at 62 °C, 1 min at 70 °C; 5 min at 70 °C; hold at 4 °C.
- Transfer each product from step 4.3.2 into new 1.5 mL tubes. Add 97.5 µL of magnetic beads to each tube. Mix by vortexing and incubate at RT for 5 min.
- Purify the products as described from step 1.6.2 to step 1.6.4.
- Elute each purified product as described in steps 1.6.5 and 1.6.6 by adding 25 µL of low-TE buffer.
5. Quality control and dilution of the DNA library
- Quantify each prepared DNA library from step 4.3.5 by the fluorometer assay according to the manufacturer’s manual using 2 µL of DNA library as starting material.
- The accepted concentration of the DNA library is ≥ 0.5 ng/µL and that of the positive control (Table of Materials) is ≤ 15 ng/µL. If the concentration of the positive control varies too much from 15 ng/µL, repeat the quantification of the positive control until the concentration is close to 15 ng/µL. If the concentration of the library is below 0.5 ng/µL, restart from fragmentation (section 3).
NOTE: Ensure that the concentration of the positive control reaches the accepted value before quantifying the DNA library. - Dilute each library to 100 pmol by adding nuclease-free water. Add 1 µL of library to n µL of nuclease-free water; calculate n using the equation below:
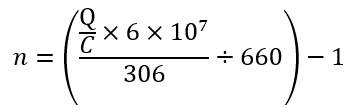
where Q is the concentration of each library measured by the fluorometer assay and C is the concentration of the positive control measured by the fluorometer assay.
6. Sequencing
- Prior to start, prepare 48 µL (with a 20% excess) of 1 M NaOH for each sample and one nuclease-free 1.5 mL tube. Thaw the master mix PCR buffer (Table of Materials) (2000 µL in volume) at RT. Bring the sphere particles (Table of Materials) to RT.
- Library pooling
- Vortex each diluted library from step 5.3 and briefly spin 4x on a mini centrifuge for 3 s each time. Take 5 µL of each library to pool into the nuclease-free 1.5 mL tube. Vortex the mixed library and briefly spin on a mini centrifuge for 3 s.
- Emulsion PCR using an emulsion system
- Add 150 µL of breaking solution to 2 new recovery tubes (Table of Materials). Install the new recovery tubes, recovery router and amplification plate.
- Mix by inverting the oil bottle (Table of Materials) 3 times. Ensure that both the oil and recovery solution (Table of Materials) are at least 2/3 full.
- Vortex the master mix PCR buffer for 30 s and briefly spin on a mini centrifuge for 3 s. Vortex the sphere particles and the mixed library from step 6.2.1 for 1 min and briefly spin on a mini centrifuge for 3 s.
- Prepare a 2400 µL ligation mix by adding 172 µL of nuclease-free water, 8 µL of mixed library from step 6.3.3, 120 µL of enzyme mix (Table of Materials), and 100 µL of sphere particles to the tube containing 2000 µL master mix PCR buffer.
- Set a pipette to 800 µL. Load the ligation mix from step 6.3.4 to the reaction filter (Table of Materials) through the sample port. Use a 1000P pipette to add 200 µL of reaction oil to the reaction filter.
- Select the program Proton: Ion PI Hi-Q OT2 200 Kit, and then, select Assisted button to ensure that the device has been set up correctly by following the instructions on the monitor. Then, click Next to start the program.
- Enrichment by an automatic enrichment system
- When the emulsion PCR program is completed, click Next, and then click Final Spin to spin for 10 min. Take out the 2 recovery tubes after clicking Open Lid.
- Discard the supernatant from the 2 recovery tubes until 100 µL remains in each tube, and label accordingly. Mix the solution well and transfer to a new 1.5 mL tube.
- Add 200 μL of nuclease-free water to each recovery tube, wash by pipetting up and down several times, and transfer all the solution to the 1.5 mL tube in step 6.4.2. Repeat the wash step once.
- Add 200 µL of nuclease-free water to one of the recovery tubes and wash by pipetting up and down several times. Transfer all the solution to the other recovery tube and wash by pipetting up and down several times. Then, transfer all the solution to the same 1.5 mL tube from step 6.4.3. Vortex the 1.5 mL tube for 30 s and centrifuge for 8 min at 15,500 x g.
NOTE: The final total volume of the emulsion PCR product in this step should be approximately 1200 µL. - Discard the supernatant in the tube and keep 20 µL of the emulsion PCR product. Add 80 µL of resuspension solution (Table of Materials) to the tube. Mix by pipetting up and down.
- Prepare a 320 µL melt-off solution for each chip by adding 280 µL of polyethylene glycol sorbitan monolaurate solution (Table of Materials) and 40 µL of 1 M NaOH.
NOTE: 1 M NaOH should be stored at 4 °C or freshly prepared. Vortex before use. - Vortex the tube containing C1 beads (Table of Materials) for 30 s. Take 100 µL of C1 beads to a new 1.5 mL tube. Place the 1.5 mL tube onto the magnetic stand for 2 min at RT. Discard all the supernatant after the beads have settled without disturbing the beads.
- Add 1 mL of wash solution C1 (Table of Materials) to the tube from step 6.4.7. Vortex for 30 s. Place the tube onto the magnetic stand for 2 min at RT. Discard all the supernatant after the beads have settled without disturbing the beads. Resuspend the beads by adding 130 µL of bead capture solution (Table of Materials).
- Enrichment system (ES) setup
- Load the sample (100 µL emulsion PCR product) from step 6.4.5, the washed beads (130 µL) from step 6.4.8, ES wash solution (300 µL) (Table of Materials), and melt-off solution (300 µL) from step 6.4.6 into the 8-tube strip. The layout order is: sample (tube 1), washed beads (tube 2), ES wash solution (tubes 3, 4, 5), and melt-off solution (tube 7). Keep tubes 6 and 8 empty.
- Place the 8-tube strip from step 6.4.9.1 onto the ES. Install a pipette tip and a new 0.2 mL tube and start the program.
NOTE: Ensure that pipetting works normal.
- Wash the sphere particles after the enrichment is completed.
- Centrifuge the 0.2 mL tube from step 6.4.9.2 for 5 min at 15,500 x g. Discard the supernatant and keep 10 µL of the enrichment product. Add 200 µL of nuclease-free water to the tube. Mix by vortexing.
- Centrifuge the 0.2 mL tube from for 5 min at 15,500 x g. Discard the supernatant and keep 10 µL of the enrichment product. Add 90 µL of nuclease-free water to the tube. Mix by vortexing.
- Template preparation
- Vortex the positive control and spin briefly. Add 5 µL of positive control to the 100 µL template (the enrichment product from step 6.4.10.2). Vortex and centrifuge for 5 min at 15,500 x g. Discard the supernatant and keep 10 µL of the template.
- Add 20 µL of sequencing primer (Table of Materials) and 15 µL of annealing buffer (Table of Materials) to the template tube from step 6.5.1. Vortex the tube and briefly spin on a mini centrifuge for 3 s.
- Incubate the tube from step 6.5.2 in the thermal cycler with heated lid. Run the program with the following settings: 2 min at 95 °C, 2 min at 37 °C, hold at 4 °C.
- Add 10 µL of loading buffer (Table of Materials) to the tube from step 6.5.3. Mix by pipetting up and down.
- Sequencer initialization
- Check the tank pressure of nitrogen gas (total pressure ≥ 500 psi, output pressure ≥ 10 psi, optimum 20-30 psi). Top-up 100 mL deionized water (18.2 MΩ) to C1 and C2 tubes (Table of Materials) and install them to corresponding C1 and C2 positions on the sequencer.
- Prepare W1 (32 µL of 1 M NaOH) and W3 (40−50 mL of W3 buffer [Table of Materials]) solutions. Prepare W2 solution by adding 1920 mL of deionized water (18.2 MΩ), a whole bottle of W2 buffer (Table of Materials) and 8−12 µL of 1 M NaOH, and invert 4−8 times to mix.
NOTE: Because the water quality varies geologically, adjust the volume of 1 M NaOH as needed. The starting pH of W2 is 5.9−6.1, and the optimal range after adjustment is 7.4−7.6. Change and install new reagent tubes and use lately used chip for washing. - Prepare the 4 new empty tubes from the sequencing supplement kit (Table of Materials). Label the 4 tubes as dGTP, dCTP, dATP, and dTTP, and add 70 µL of dGTP, dCTP, dATP, or dTTP (Table of Materials) to the corresponding tube (i.e., 70 µL dGTP to the tube labeled as dGTP, etc.). Vortex the tubes before use. Install the tubes to the corresponding positions designated on the sequencer (Table of Materials).
- Chip wash
- Wash the chip (Table of Materials) once by injecting 100 µL of isopropanol into the loading well of the chip. Remove the expelled liquid from the opposite well.
- Wash the chip twice by injecting 100 µL of nuclease-free water into the loading well of the chip. Remove the expelled liquid from the opposite well.
- Wash the chip once by injecting 100 µL of 0.1 M NaOH into the loading well of the chip. Remove the expelled liquid from the opposite well. Incubate at RT for 1 min.
- Wash the chip once by injecting 100 µL of nuclease-free water into the loading well of the chip. Remove the expelled liquid from the opposite well.
- Wash the chip twice by injecting 100 µL of isopropanol into the loading well of the chip. Remove the expelled liquid from the opposite well. Dry by blowing nitrogen onto the chip. Keep away from light.
- Sample loading and sequencing
- Mix the 55 µL sample from step 6.5.4 by pipetting up and down and load the sample to the loading well of the chip.
NOTE: Keep the pipette tip and the 0.2 mL PCR tube used in this step. - Place the chip onto the chip mini centrifuge (Table of Materials) when
- Prepare two new 1.5 mL tubes for the annealing buffer and flushing solution. Prepare the 50% annealing buffer by adding 500 µL of annealing buffer and 500 µL of nuclease-free water. Prepare the flushing solution by adding 500 µL of annealing buffer and 500 µL of 100 % 2-propanol.
- Prepare two new 1.5 mL tubes and prepare the foaming mixture by mixing 49 µL of 50% annealing buffer and 1 µL of foaming solution (Table of Materials) in both tubes.
- Set a pipette to 100 µL. Make bubbles by pipetting air into the foaming mixture from one of the two tubes from step 6.8.4. Make > 120 µL of bubbles and keep pipetting until no outstanding visible bubbles can be seen. Load 120 µL of bubbles into the loading well.
NOTE: Ensure that there are no outstanding visible bubbles. Otherwise, start it over. - Transfer the excessive expelled liquid from the exit well from step 6.8.5 to the loading well by pipetting. Do not pipette bubbles. Centrifuge the chip for 30 s on the chip mini centrifuge.
- Repeat step 6.8.5 by using the second tube containing the foaming mixture from step 6.8.4.
- Add 55 µL of the 50% annealing buffer to the 0.2 mL tube kept in step 6.8.1. Use the kept pipette tip in step 6.8.1 to pipette up and down. Load all the 55 µL annealing buffer to the loading well. Centrifuge the chip for 30 s on the designated chip mini centrifuge.
- Load 100 µL of flushing solution into the chip loading well and discard the expelled liquid from exit well. Repeat this loading step once.
NOTE: If there are bubbles in the chip, expel small bubbles by big bubbles and flush by flushing solution. This can be achieved by pipetting 100 µL of flushing solution and leaving 5 µL of air below the flushing solution. Therefore, when pipetting the 105 µL into the chip, air will form a big bubble that can expel the small bubbles, and then, the big bubble can be expelled by the following flushing solution. - Load 100 µL of the 50% annealing buffer into the chip loading well. Repeat this loading step for a total of 3 times.
- Add 6 µL of the sequencing enzyme into 60 µL of the 50% annealing buffer into a new 1.5 mL tube. Mix by pipetting up and down. Load 65 µL of this mixed solution into the chip loading well. Pipette slowly to avoid foaming.
- Keep the chip away from light and incubate at RT for 5 min.
- After the incubation, immediately load the chip onto the sequencer and click Start the sequencing run on the screen to start sequencing.
NOTE: The sequencing raw data and quality control files will be uploaded automatically to the company for data analysis.
- Mix the 55 µL sample from step 6.5.4 by pipetting up and down and load the sample to the loading well of the chip.
Based on this modified protocol, the semiconductor sequencing platform was for the first time, applied for PGT-A. We tested on biopsies from both cleavage-stage blastomeres and blastocyst-stage embryos. It is suggested that the biopsied cells undergo WGA as soon as possible to prevent any degradation of DNA. A previous study compared the performance of different WGA methods and indicated that the method we described here had the best uniformity at the bin size of 100 KB20. Considering the performance of both uniformity and median absolute pairwise difference (MAPD)21, this WGA method was chosen for PGT-A using the semiconductor sequencer. Through a retrospective statistical analysis on 186 cleavage stage and 1135 blastocyst stage embryos, we observed that the WGA success rates were 95.4% in blastomere samples and 96.9% in blastocyst samples (Figure 1). The purification step before library construction as a size selection procedure was crucial for sequencing quality by capturing large DNA fragments. Additionally, it facilitated the input amount of 300 ng for library construction. The enzymatic fragmentation method enabled an efficient shearing of WGA products into approximately 160 bp.
Data analysis was conducted using the Euclidean distance and circular binary segmentation (EDCBS) analysis system. In-house validation was performed to evaluate the robustness of this bioinformatic algorithm. We established a reference database exclusive for PGT-A through sequencing 379 WGA products from 66 cell lines with known karyotypes by analyzing a bin size of 100 KB. From this database, a reference range was delineated as the threshold for copy number variant (CNV) calling and 10 MB was set as the cutoff for the detection level. Both sensitivity and specificity reached over 99% at this threshold (Table 1). In the application for PGT-A on embryo biopsies, the window size was set to 400 KB with a sliding window approach to reach enough reads. Quality control (QC) of each sample was determined by unique reads, MAPD and standard deviation of copy number variant (CNV∙SD). Sample beyond one of the three indexes was defined as QC failure (Figure 2C). Interpretation of chromosome scatter plots (Figure 2A,B,D) was conducted by qualified geneticists following a workflow by comparing the identified CNV to DECIPHER, DGV, or ClinGen databases. Individual discrepancies were controlled by an expert curation procedure. Chromosomal abnormalities were grouped into aneuploidy and mosaicism in blastocyst samples. A copy number gain or loss within the range of 30%−70% was classified as carrying mosaic chromosomal composition; otherwise, the result would be interpreted as either euploidy or aneuploidy. In this study, the euploid rates were 45.2% in blastomeres and 52.3% in blastocysts, which echoed to published data22,23.
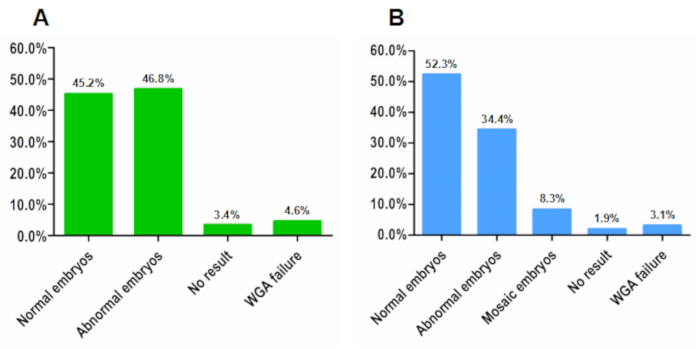
Figure 1: Demographic statistics of 1321 embryo biopsies tested by this method. (A) Data from 186 cleavage-stage embryos. (B) Data from 1135 blastocyst-stage embryos. WGA success rates are over 95% in both types of specimens. Sequencing quality control failure rates are 3.4% in the cleavage-stage group and only 1.9% in the blastocyst-stage group. Please click here to view a larger version of this figure.
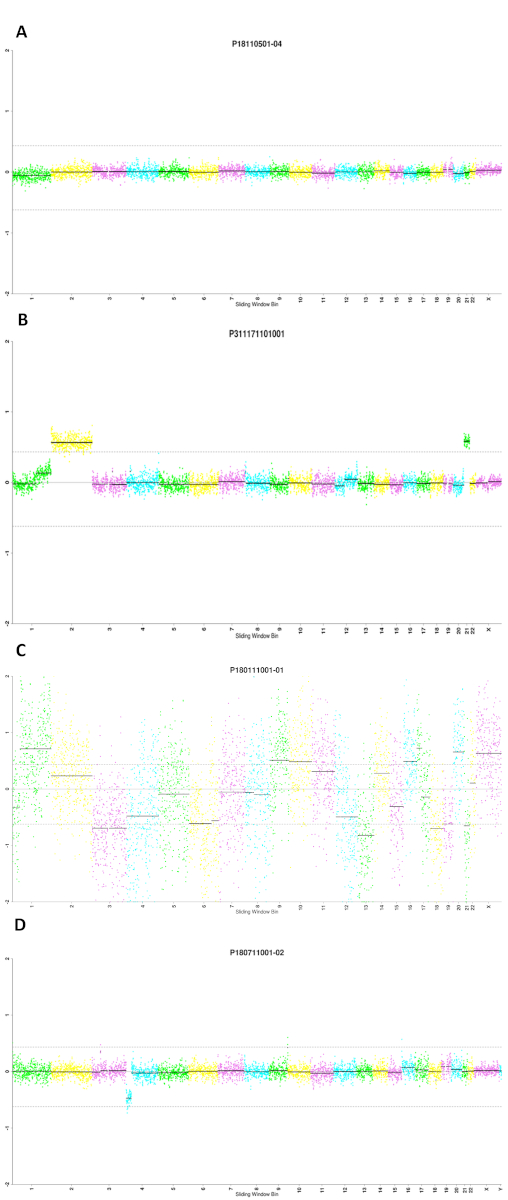
Figure 2: Representative results of PGT-A clinical application of embryo for the 23 pairs of chromosomes. Representative results of (A) euploidy; (B) aneuploidy (seq[GRCh37] (2)x3, (21)x3); (C) sequencing QC failed sample due to CNV∙SD at 0.6571 (acceptance ≤ 0.4); (D) segmental mosaic deletion (55%) of 4p16.3p15.1 (29.50 MB). Please click here to view a larger version of this figure.
| CNV threshold value | ||||||
| (-0.23, 0.20) | (-0.32, 0.26) | (-0.41, 0.32) | (-0.51, 0.37) | (-0.62, 0.43) | (-0.73, 0.48) | |
| Sensitivity | 100.00% | 100.00% | 100.00% | 100.00% | 98.30% | 96.02% |
| Specificity | 72.41% | 81.03% | 91.38% | 99.10% | 99.54% | 100.00% |
Table 1: Sensitivity and specificity between different Log R ratios by the semiconductor sequencer. A total of 240 samples with known euploid karyotype results were tested by this method and called at different Log R ratios.
