The four steps of in vitro transcribed RNA-based luciferase reporter assay: PCR to generate DNA template for In vitro transcription, in vitro transcription to generate mRNA, mRNA transfection, and luciferase measurement, can be seen in the schematic diagram (Figure 1). Designing of primers for both DNA templates (Fluc and Rluc) and the general scheme of overhang extension PCR is illustrated in the schematic (Figure 2A). After PCR, the correct sized PCR product was detected by TAE agarose gel electrophoresis (Figure 2B). Subsequently, the PCR product is used as the template to synthesize RNA in vitro (Figure 3A), which is purified and run in TBE gel electrophoresis to verify the size (Figure 3B). The purified and verified mRNA is transfected into cells using cationic lipid transfection reagent (Figure 3C). Primers used in this protocol are listed in Table 6.
The in vitro transcribed RNA-based luciferase reporter assay was developed to understand the role of 5'-poly(A) leader in mRNA translation during poxvirus infection. Using this assay, we tested the translation efficiency of a Fluc mRNA that contains a 5'-poly(A) leader (12 nt) in uninfected and VACV-infected cells. The Fluc value was normalized using Rluc value in both uninfected and VACV-infected cells to determine the relative Fluc activity (i.e. Fluc activity/Rluc activity) (Figure 4A). The division of Fluc by Rluc normalized the transfection efficiency and RNA stability in a particular well. Using this analysis approach, we determined that a 5'-poly(A) leader containing mRNA has a translational advantage during VACV infection (Figure 4B). The advantage in infected cells was not due to differential transfection efficiency or mRNA stability as the RNA level was similar in uninfected and VACV infected cells 5 h post mRNA transfection19.
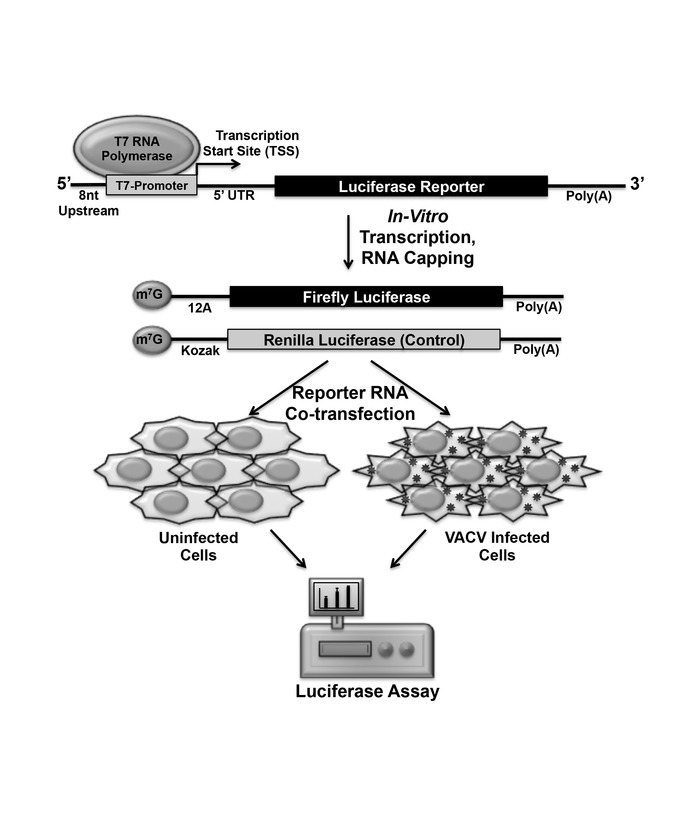
Figure 1: Schematic of the experimental procedure. PCR is used to generate a DNA template with the desired elements. mRNA encoding a luciferase reporter gene is synthesized in vitro using a T7 RNA polymerase-based system. A Firefly luciferase (Fluc) mRNA is co-transfected with a Renilla luciferase (Rluc) mRNA into uninfected or VACV-infected cells. Luciferase activities are measured using a luminometer with dual luciferase capability. Please click here to view a larger version of this figure.
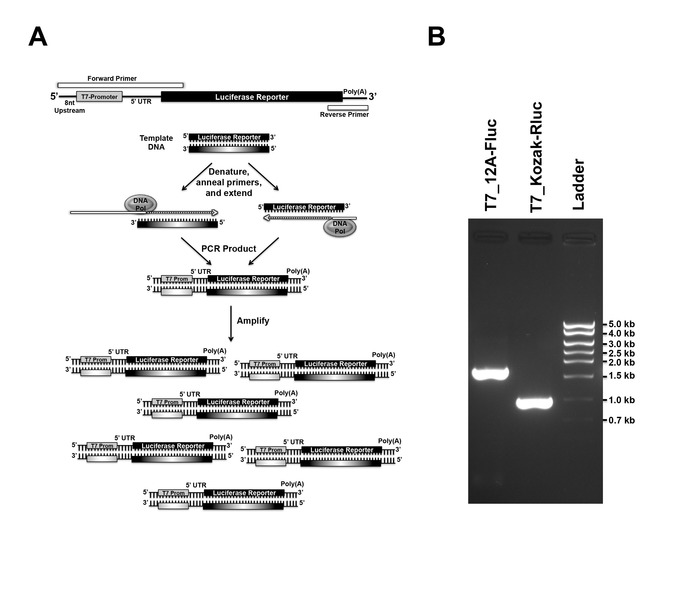
Figure 2: Primer design and PCR-based DNA amplification. (A) The forward primer is synthesized to include an 8nt random sequence, T7 promoter followed by a desired 5'-UTR and part of the 5' end of the luciferase reporter gene, while the reverse primer includes a T-tract to generate poly(A) tail and the 3' end of the luciferase reporter gene. By overhang extension PCR using a plasmid template containing a luciferase gene, a DNA template is generated. (B) DNA band of the desired size from PCR reaction was detected using 1% agarose TAE gel electrophoresis. Please click here to view a larger version of this figure.
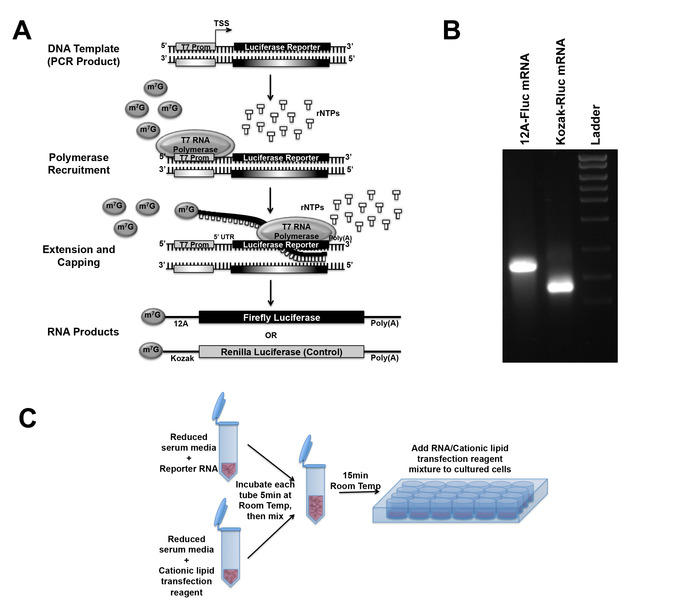
Figure 3: mRNA synthesis and transfection. (A) Schematic of in vitro transcription. DNA amplified by PCR containing the luciferase gene downstream from the 5'-UTR of interest and the T7 promoter is used as the template. The T7 RNA polymerase is recruited to the promoter and adds ribonucleotides, shown in white, from 5' to 3' direction. Once mRNA is 25-30 nt long, m7G cap is added using an anti-reverse cap analog, ARCA. (B) RNA bands from in vitro transcription ware detected using 1.5% agarose TBE gel electrophoresis. (C) Schematic demonstrating the transfection of reporter mRNA into cells. Medium containing either the reporter mRNA or cationic lipid transfection reagent in separate tubes is allowed to equilibrate at room temperature for 5 min. The solutions are then mixed followed by incubation at room temperature for 15 min after which the RNA/transfection reagent mixture is added into cells in culture plates. Please click here to view a larger version of this figure.
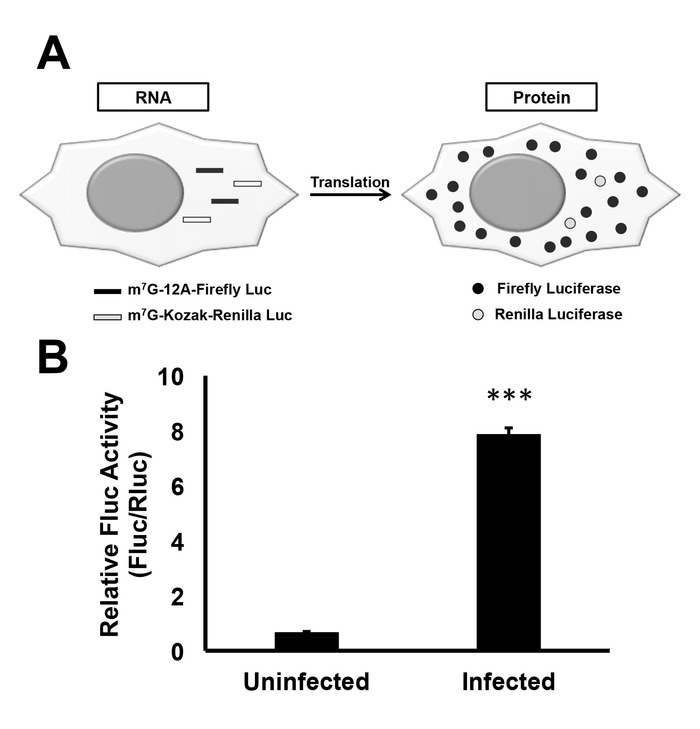
Figure 4: Increased translational efficiency of mRNA containing a 5'-poly(A) leader. (A) Fluc mRNA containing a poly(A) leader in the 5'-UTR and Rluc mRNA with the Kozak consensus sequence in the 5'-UTR are co-transfected into cells. (B) Fluc mRNA with 5'-poly(A) leader was transfected in uninfected and VACV-infected cells along with Rluc mRNA. 5 hpi, luciferase activity was measured using a luminometer. Rluc normalized Fluc activity is represented in uninfected and VACV-infected cells. Error bars indicate the standard deviations (SD) of at least three repeats. Student's t-test was used to determine P-values; ***P-value < 0.001. Please click here to view a larger version of this figure.
| Elements | Sequence |
| T7 Promoter | TAATACGACTCACTATAGGG |
| Poly(A) leader | AAAAAAAAAAAA, ranging from 3 to 51 As |
| Kozak sequence | GCCACC |
| Poly(A) tail | AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA…… |
Table 1: Sequences used in the method – the table contains the sequences of the T7 promoter, poly(A) leader, Kozak sequence, poly(A) tail.
| Components | Volume |
| DNase free water: | 38 µL |
| 2X High-fidelity DNA polymerase Master mix: | 50 µL |
| Forward Primer (10 µM): | 4 µl |
| Reverse Primer (10 µM): | 4 µl |
| Luciferase DNA Template (1-10 ng/µL): | 4 µl |
| Total: | 100 µL |
| Source for Fluc DNA template is pGL3-Fluc plasmid | |
| Source for Rluc DNA template is pRL-Rluc plasmid |
Table 2: PCR reaction – the order and the volume of components added in the PCR reaction.
| Step | Temperature | Time | Cycle |
| Initial denaturation | 95 °C | 2 min | (1x Cycle) |
| Denaturation | 95 °C: | 15 s | |
| Annealing | X °C: | 30 s | (25x Cycle) |
| Extension | 72 °C: | T min | |
| Final Extension | 72 °C: | 7 min | (1x Cycle) |
| Hold | 4 °C: | ∞ |
Table 3: PCR Program – the steps for PCR program along with temperature, time and cycle.
| Components | Volume | |
| DNase-RNase free water: | up to 20 µL | |
| NTP Buffer Mix (20 mM of each rNTP): | 2 µL | |
| Cap Analog (40 mM): | 4 µL | |
| Template PCR Product (400 ng)*: | X µL | (PCR Product Concentration dependent) |
| T7-RNA polymerase Mix: | 2 µL | |
| Total: | 20 µL | |
| *T7_12A-Fluc and T7_Kozak-Rluc template used to synthesize 12A-Fluc and Kozak-Rluc mRNA, respectively. |
Table 4: In vitro transcription reaction – the order and the volume of components added for in vitro transcription reaction.
| Steps | Volume/Time |
| Inject Luciferase Assay Substrate (Fluc): | 30 µL |
| Wait / Incubation time: | 2 s |
| Luminescence Measurement (Fluc): | 10 s |
| Stop & Glo Substrate (Rluc): | 30 µL |
| Wait / Incubation time: | 2 s |
| Luminescence Measurement (Rluc): | 10 s |
Table 5: Luciferase Measurement Settings – the steps for luciferase measurement with the recommended volume or time.
| Primers | Sequence |
| T7-12A-Fluc Forward | ATCGACGATAATACGACTCACTATAGGGaaaaaaaaaaaa ATGGAAGACGCCAAAAACATAAAG |
| T7-12A-Fluc Reverse | TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTACACGGCGATCTTTCCGC |
| T7-Kozak-Rluc Forward | ATCGACGATAATACGACTCACTATAGGGatcgtagccacc ATGACTTCGAAAGTTTATGATC |
| T7-Kozak-Rluc Reverse | TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTATTGTTCATTTTT GAGAACTCGCTC |
Table 6: Primers – the primers used in this method with complete sequences.
Supplementary Figure 1: Analysis of raw data – steps for analyzing raw data to get normalized data. Please click here to download this file.
