Structural Properties of Binding Motifs
Representative binding motifs for the four different partial-charge sets are depicted in Figure 2, after 15 ps of MD. In Figure 2a, for the (above-described) literature-derived charges, it can be seen that there is a prominent hydrogen-bonding interaction with a surface proton. From careful analyses of the trajectory, the hydrogen bonds are mostly surface-proton-bound whilst the other three (AIMD-derived)20 charge sets do not feature such a strong Coulombic interaction with a surface proton. Referring to Figure 1f of ref. 20, which depicts the AIMD-relaxed dye-binding configuration after ~8.5 ps, there is also less evidence of strong hydrogen-bonding with a surface proton, so this is qualitatively consistent in tailoring the present forcefield-based MD with AIMD-based charge sets to achieve qualitatively similar substrate-binding arrangements (Figure 2b-d). Indeed, the smaller magnitude of the partial charges in the literature-derived case vis-à-vis those sampled in various ways from AIMD (Table 1 & Table 2) leads to a lesser extent of charge shielding in comparison to larger-magnitude partial RTIL charges, which serves to emphasize more the electrostatic (hydrogen-bond) interaction with the surface proton evident in Figure 2a. In any event, interestingly, the Mulliken-derived charge set shows a certain sustained 'kinking' of the dye to have a prominent hydrogen bond with a bridging oxygen atom at the anatase surface (Figure 2b), which is redolent of the PBE system of ref. 15 without Grimme dispersion (Figure 2d therein): the generally-recognized inferior quality of Mulliken charges leads to this less physical, persistent kinking, which has been studied in more detail in refs. 12, 13 & 20. Tellingly, the better-quality charge fits (EHT and Hückel) from AIMD20 lead to more realistic N719-binding motifs in Figure 2c,d, which are in accord with PBE-based BOMD featuring Grimmer-D3 dispersion in ref. 20 (cf. Figure 1f therein); comparison of motifs with ref. 20 shows this is somewhat more the case for Hirshfeld charges.
Empirical-Potential Spectra
Having established at gross structural level already the clear influence of RTIL partial-charge sets in parameterizing and tailoring pragmatically the efficacy of forcefield-based MD vis-à-vis the best-quality AIMD data available, we now turn to considering empirical-potential-based MD for replication of N719 vibrational spectra. The mass-weighted VACF spectra for the four different parametrized charge sets are shown in Figure 3; as mentioned previously, although all four MD-generated spectra have the same forcefields for their dyes and surfaces, they differ in the partial charges applied to the RTIL cations and anions.
Now, prior to discussing the MD-predicted vibrational spectra, let us make some brief explanatory comments about their more fundamental nature and higher-level interpretation. The continuous colored lines in Figure 3 denote the (empirical-potential) MD-based spectra in the range from 0 to 2500 cm-1, for all four RTIL partial-charge sets. The dashed vertical gray lines are established experimental modes for the N719 dye and are at the frequencies 1230, 1380, 1450, 1540, 1600, 1720 and 2100 cm-1, respectively36. The two grey spectral insets are experimental ATR-FTIR results from ref. 37, with the topmost one being the spectra for unsolvated N719 powder and the bottom one that for unsolvated N719 powder adsorbed on anatase. These experimental results are intended as a guide only, in that the spectra are slightly different in other studies and the two experimental insets are themselves somewhat different due to adsorption to anatase. A solvent's presence would be expected to alter the spectra, whether it be an RTIL or the more traditional acetonitrile. Also, it must be noted that the experimental spectra have a reduced frequency window available, and are in effect denoting a mixture of dynamical properties of multiple dyes in various geometries; in contrast, it ought to be borne in mind that the results are for a single N719 molecule adsorbed to the anatase substrate, therefore leading to an inevitably more sharp signal.
Now, the modes themselves are discussed more in refs. 14 & 18; the present discussion focusses more on fidelity of each technique in reproducing these, as opposed to their underlying nature.
0-500 cm-1: The previous AIMD results (i.e., PBE-based BOMD with Grimme-D3 dispersion and explicit RTIL solvation)20 show a cluster of spectroscopic peaks in the 300-400 cm-1 region. In terms of closeness to the ab initio spectra, the classical charge sets were ranked in order from closest to furthest away as EHT, Hirshfeld, literature-based and Mulliken.
500-1000 cm-1: The ab initio MD spectra20 display prominent vibrational peaks at 625, 750 and 825 cm-1; the main peaks present in the classical spectra are at 600 and 800 cm-1 for the literature-derived charges, 525 and 800 cm-1 for the Mulliken charges, 675, 810, and 900 cm-1 for the EHT charges and 650,800 and 900 cm-1 for the Hirshfeld charge set. Although the literature and Mulliken charge sets reproduce grosso modo some of the features of the ab initio spectra, both the EHT and Hirshfeld-derived charge sets generate spectra which have their main peaks only 25-75 cm-1 above those of the ab initio ones. Given the much closer structural resemblance of the binding motif in Figure 2c,d relative to Figure 1f of ref. 20, this excellent, semi-quantitative agreement is encouraging for tailoring and optimising pragmatically forcefields using high-quality AIMD.
1000-1500 cm-1: The BOMD spectra evince strong peaks at 1000, 1300, and 1400 cm-1 20, whereas the main peaks present in the forcefield-based spectra are at 1075 and 1200 cm-1 for the literature-derived charges, 1080, 1350 and 1450 cm-1 for Mulliken charges, 1075 and 1200 cm-1 for the EHT charges and 1075 and 1250 cm-1 for the Hirshfeld charge set. Although the forcefield-based RTIL-charge sets produce results close to those of AIMD simulation20, a key difference between these and BOMD lies in the important fact that the present empirical-potential simulations allow only for physical adsorption rather than also for the possibility of chemical adsorption. Clearly, this will have a particularly strong effect in this vibrational-bending spectral region and serves to explain a good deal of the altering of vibrational modes related to surface binding.
1500+ cm-1: In the stretch-dominated frequency range above 1500 cm-1, the solvated ab initio spectra20 exhibit modes at 1525, 1575, 1600, 1700 and 2075 cm-1. All four forcefield-based spectra have modes in the region of 1525 cm-1, whilst none capture the thiocyano mode around 2075 cm-1. The literature-derived charge set produces spectra which shows vibrational modes at 1625 and 1700 cm-1, whereas spectra derived from the use of Mulliken charges result in modes at 1600, 1675 and 1775 cm-1. The EHT-generated spectra have a mode at 1700 cm-1, and the Hirshfeld-generated spectra does a rather excellent job of reproducing DFT-based MD results with modes at 1575, 1600 and 1700 cm-1. It is clear that the greater sophistication of the Hirshfeld charge fitting does pay important dividends with respect to basic qualitative reproduction of salient spectral features, showcasing further the important effect of RTILs and suitable treatment of their electrostatics on capturing the essential details of N719 vibrational properties.
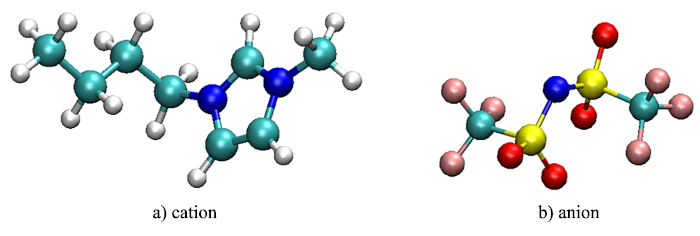
Figure 1: Cation and anion images of the considered system. Carbon is shown in cyan, nitrogen in blue, oxygen in red, hydrogen in white, sulphur in yellow, and fluorine in pink. Please click here to view a larger version of this figure.
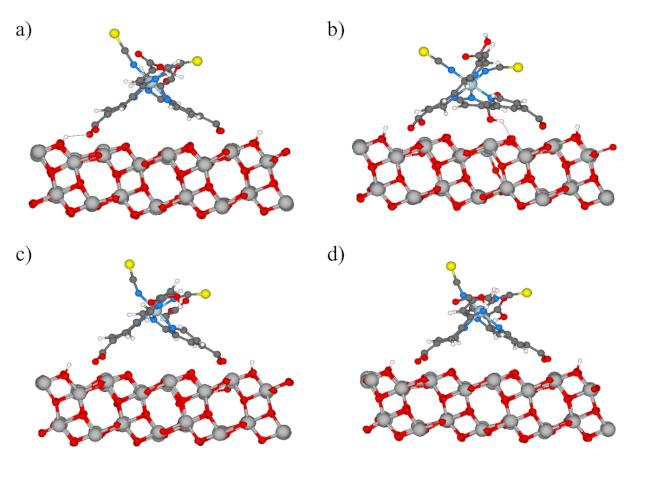
Figure 2: Frontal view showing the relaxed geometries of the systems under consideration, after 15 ps of MD. Carbon is shown in grey, nitrogen in blue, oxygen in red, hydrogen in white, titanium in silver, Sulphur in yellow and ruthenium in light green. The explicitly solvated systems are shown without RTIL ions for ease of viewing. Please click here to view a larger version of this figure.
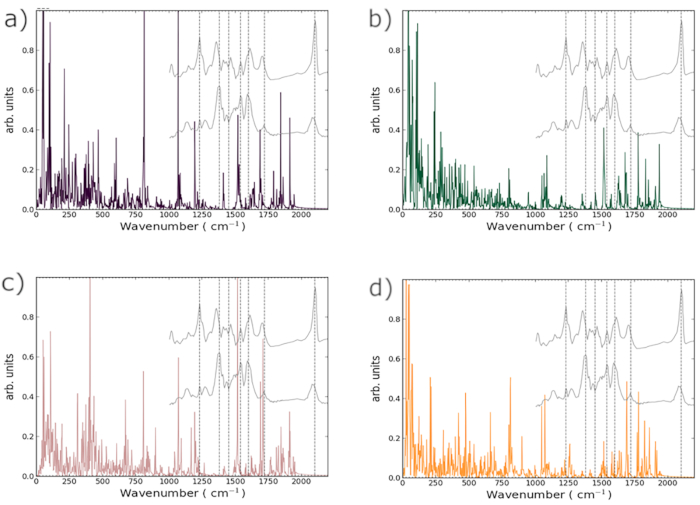
Figure 3: Vibrational Spectra for adsorbed-N719 from MD; the forcefield-based MD simulations differ from each other only in their partial-charge parametrization of the RTIL. Within each plot the grey (lower) / (upper) inset corresponds to the experimental ATR-FTIR signal from León37, for (dry-N719 adsorbed onto anatase) / (dry-N719 powder). The dashed lines indicate established vibrational modes30. (a) Literature-derived RTIL Charges, (b) Mulliken RTIL Charges, (c) Extended-Huckel-Theory Charges, and (d) Hirshfeld RTIL Charges. Please click here to view a larger version of this figure.
| Atom | Anion | |||
| Literature | Mulliken | EHT | Hirshfeld | |
| N | 0.15 | 0.005 | -0.76 | -0.1 |
| C | -0.11 | -0.06 | 0.65 | 0.14 |
| N | 0.15 | 0.005 | -0.76 | -0.1 |
| C | -0.13 | -0.06 | 0.2 | 0.14 |
| C | -0.13 | -0.06 | 0.2 | 0.14 |
| C | -0.17 | -0.34 | 0.274 | 0 |
| H | 0.21 | 0.2 | 0.15 | 0.11 |
| C | -0.17 | -0.34 | 0.354 | 0 |
| H | 0.21 | 0.18 | 0.15 | 0.09 |
| H | 0.21 | 0.18 | 0.15 | 0.09 |
| H | 0.13 | 0.18 | 0.08 | 0.08 |
| H | 0.13 | 0.18 | 0.08 | 0.08 |
| H | 0.13 | 0.18 | 0.08 | 0.08 |
| C | 0 | -0.25 | -0.16 | 0 |
| H | 0.13 | 0.18 | 0.08 | 0.08 |
| H | 0.13 | 0.18 | 0.08 | 0.08 |
| C | 0 | -0.25 | -0.16 | 0 |
| H | 0.045 | 0.151 | 0.08 | 0.04 |
| H | 0.045 | 0.151 | 0.08 | 0.04 |
| C | -0.17 | -0.34 | -0.24 | 0 |
| H | 0.045 | 0.151 | 0.08 | 0.04 |
| H | 0.045 | 0.151 | 0.08 | 0.04 |
| H | 0.045 | 0.151 | 0.08 | 0.04 |
| H | 0.045 | 0.151 | 0.08 | 0.04 |
| H | 0.045 | 0.151 | 0.08 | 0.04 |
| mean modification | 0.0062 | 0.0159 | — | 0.0195 |
Table 1: Different parametrized charge sets for the atomic partial charges of the cation. Mean modification is the per atom alteration of charge necessary to achieve overall charge neutrality.
| Atom | Anion | |||
| Literature | Mulliken | EHT | Hirshfeld | |
| N | -0.368 | -0.44 | -0.368 | -0.62 |
| S | 1.311 | 0.5 | 1.311 | 1.41 |
| O | -0.717 | -0.3 | -0.717 | -0.64 |
| C | 1.09 | 0.25 | 1.09 | 0.8 |
| mean modification | — | 0.0062 | — | 0.0045 |
Table 2: The different parameterized charge sets for the atomic partial charges of the anion. Mean modification is the per atom alteration of charge necessary to achieve overall charge neutrality.
