1. Primary screening: cell culture and plate seeding
- To prepare poly-L-lysine (PLL)-coated plates, dispense 20 μL/well of a 25 μg/mL stock solution of PLL in white or black 384-well optical bottom plates using an electronic multichannel pipette or a reagent dispenser. Incubate the plates at room temperature for 0.5–2 h.
NOTE: If using the black 384-well plates, expect the background signal to be lower compared to the white plates. Black plates are recommended to reduce bleed-through of luminescence between adjacent wells. - To preserve the coated plates and wash off the excess PLL, remove the PLL by flicking it over the sink, tap dry over a paper towel, and add 40 μL/well of diluted 1x solution of antibiotic-antimycotic using an electronic multichannel pipette or a reagent dispenser. Store PLL-coated plates at 4 °C until ready for plate seeding.
- Maintain HTLA cells (kindly provided by Dr. Richard Axel)–a human embryonic kidney cell line (HEK293T) stably expressing β-arrestin2-TEV and tTA-driven luciferase–in complete Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% of Fetal Bovine Serum, 5% of Bovine Calf Serum, 2.5 μg/mL of puromycin, 50 μg/mL of hygromycin, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified incubator containing 5% CO2.
- Culture HTLA cells in 150 mm dishes and pass cells twice a week at a dilution factor of 1:10, with optimal cell passage number of 5–25. Ensure that a sufficient number of 150 mm dishes are confluent the day of 384-well plate seeding, depending on the scale of the primary screen.
NOTE: Usage of HTLA cells greater than passage 25 may result in reduced viability, yielding suboptimal results. - To seed HTLA cells for the primary screen, gently rinse the confluent 150 mm dish(es) with 1x phosphate-buffered saline (PBS), pH 7.4. Detach cells with approximately 6 mL of 0.05% Trypsin/0.53 mM EDTA, and transfer to a centrifuge tube containing at least equal amount of complete Dulbecco's modified Eagle medium (DMEM) to neutralize the trypsin.
- Spin down HTLA cells at 500 x g for 3 min and resuspend the cell pellet at a density of 0.22 x 106 cells/mL in complete DMEM, omitting the addition of 2.5 μg/mL of puromycin and 50 μg/mL of hygromycin as they can decrease transfection efficacy.
- Incubate the necessary 384-well PLL-coated plates at 37 °C to warm them before seeding cells. Remove the storage solution of 1x antibiotic-antimycotic from the 384-well PLL-coated plate(s) by flicking the plate over the sink and taping it over a paper towel to dry.
- Seed cells into the 384-well PLL-coated plates at a final density of 10,000 cells/well by dispensing 45 μL of the 0.22 x 106 cells/mL HTLA suspension using an electronic multichannel pipet. Incubate plates at 37 °C overnight. If a same-day transfection is preferred, seed cells at a density of 16,000 cells/well and perform the transfection 4 h later.
NOTE: For high transfection efficiency, 50–70% cell confluency is optimal.
2. Primary screening: DNA plate preparation and transfections
- To prepare the 384-well DNA source plate for transfection as shown in Figure 2, distribute the plasmid cDNAs encoding the GPCR-Tango constructs of interest in a 96-well plate, with a different GPCR/well. The plasmid DNA should be suspended in 0.1x Tris-EDTA (TE) buffer at a concentration of 50 ng/μL.
NOTE: The 96-well DNA plates can be sealed and stored at -20 °C, and re-used for multiple screening experiments. All cDNA encoding GPCR-Tango constructs are available commercially (see the Table of Materials) and are cloned in the pcDNA3.1 neomycin plasmid. The PRESTO-Tango GPCR Kit consists of four 96-well plates, which include 80 GPCRs each, a couple of wells with an empty vector as negative controls, and positive control wells that hold the dopamine receptor D2 (DRD2), and wells that carry a plasmid encoding a fluorescent protein (YFP) to track transfection efficiency. - Using a multichannel pipette, manually transfer DNA solution from the 96-well to the a 384-well DNA source plate, adding 10 μL per 384-well. To ensure that each condition of the experiment is assayed in quadruplicate, half of the 96-well DNA plate (rows A-D or E-H) will cover a full 384-well plate by distributing each GPCR in two quadrants (first quadrant = – compound, second quadrant = + compound), such that the same GPCR will be transfected in 8 wells of the 384-well plate (see Figure 2 as a guide).
- Assemble the following transfection reagents needed for calcium phosphate precipitation method, as described by Jordan et al.20: 0.1x TE buffer (1 mM Tris-HCl and 0.1 mM EDTA); 2.5 M CaCl2 solution; 2x Hepes buffer, pH 7.05 (50 mM HEPES, 280 mM NaCl, 1.5 mM Na2HPO4). Sterilize all the solutions by filtration and store at 4 °C. The day of transfection, allow the reagents to reach room temperature before use.
- Dilute the 2.5 M CaCl2 stock solution in 0.1x TE (1:8 dilution) to a final concentration of 0.313 M CaCl2 and vortex. Transfer 40 μL of 0.313 M CaCl2 to the 384-well DNA source plate and mix by pipetting up and down with a hand-held multichannel pipette or an automated benchtop 384-channel pipettor.
- Add 50 μL of 2x Hepes buffer to the 384-well DNA source plate, mix again by pipetting up and down and let stand for 1 min; each 384-well will have an adequate amount of DNA/transfection mixture for the transfection of nine 384-well plates, depending on the number of compounds that need to be tested. Transfer 10 μL of the DNA/transfection mixture from the 384-well DNA source plate to the seeded HTLA cells and incubate the plates overnight at 37 °C.
3. Primary screening: Cell stimulation
- Twenty-four hours later, decant the transfected cell media by gently flicking the 384-well plate over the sink and taping it over a paper towel, or with an aspirator head. Slowly add 40 μL of starving media (DMEM supplemented with 1% dialyzed fetal bovine serum (dFBS) and 1x antibiotic/antimycotic), being careful to avoid touching the cells directly.
- Pipet 20 μL of the compound of interest at a 3x concentration (final concentration of the drug in the cell plate will be 1x) into the alternating rows with (+) stimulation, and 20 μL of vehicle buffer for the alternating rows without (-) compound. Return the cell plate at 37 °C in 5% CO2 and incubate for at least 16 h.
4. Primary screening: Luminescence reading
- Prepare the Glo reagent, modified from Baker and Boyce21: 108 mM Tris–HCl; 42 mM Tris-Base, 75 mM NaCl, 3 mM MgCl2, 5 mM Dithiothrei-tol (DTT), 0.2 mM Coenzyme A, 0.14 mg/ml D-Luciferin, 1.1 mM ATP, 0.25% v/v Triton X-100, 2 mM Sodium hydrosulfite.
NOTE: Stock solutions of the reagents can be made in advance, except for D-Luciferin, which is always freshly added to the Glo reagent in its powdered form. If black plates were used, the amount of D-Luciferin can be increased up to 0.25 mg/mL. - At 16–24 h following stimulation, decant the transfected cell media by gently flicking the 384-well plate over the sink and taping it over a paper towel. Add 20 μL/well of Glo reagent and incubate the plate at room temperature for 5–20 min. Read the plates using a microplate luminescence counter, with an integration time of 1 s/well.
5. Primary screening: Data analysis
- Export the saved files from the luminescence counter as a spreadsheet; results will be recorded in relative luminescence units (RLU). Based on the layout of the 384-well plate, calculate the activation (fold change) of each receptor using the following formula:
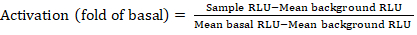
NOTE: Here Sample RLU refers to value of each of the four replicate wells of the stimulated (+ compound) quadrant, Mean background RLU is the mean of the negative controls on the plate, and Mean basal RLU refers to the mean of the untreated quadrant of that same receptor (- compound). In addition, calculate the standard deviation of the 4 data points to verify the quality of the results. It is recommended to perform a log2 transformation on the mean of the fold changes to rectify any heteroskedasticity; the log2 base is a practical choice to help identify positive hits. Empirically set the positive hit thresholds; it has to be noted that some receptors can have as low as 2-fold increase and up to 40-fold increase for others with full agonist. - Based on the results, select the GPCRs that are potential positive hits for secondary screening.
6. Secondary screening: Cell seeding and transfections
- Subculture HTLA cells in 100 mm dishes at a total cell density of 5 x 106 cells in 11 mL of complete media (4.55 x 105/mL) and incubate at 37 °C for 24 h. If a same-day transfection is preferred, seed cells at a density of 7.5 x 106 cells and perform the transfection 4 h later.
- Pre-warm the reagents needed for calcium phosphate precipitation at room temperature. Combine 450 μL of 0.1x TE buffer with 50 μL of 2.5 M CaCl2 and quickly vortex; these amounts are specific for one 100 mm dish, based on the volume of growth medium it holds.
- In a tube, add 500 μL of the TE/CaCl2 solution to 10 μg of GPCR cDNA and vortex. Add 500 µL of 2x Hepes buffer solution in the tube, shake vigorously (do not vortex), and incubate for 1 min.
NOTE: 1 µg of any plasmid encoding a fluorescent protein (e.g. YFP, mCherry, etc.) can be co-transfect with 9 µg of GPCR cDNA for a total of 10 μg. The fluorescent protein is used to track transfection efficiency, and this minimal amount will not interfere with the assay. - Immediately following the short incubation, dispense the 1 mL solution dropwise onto the cells. Gently rock the plate back and forth to evenly distribute the precipitate, taking care not to swirl the plate, and incubate at 37 °C for 24 h.
- The following day, observe the transfection efficiency by looking at the expression of the fluorescent protein under a fluorescent cell imager; transfections greater than 50% coverage are ideal.
- Incubate the necessary 384-well PLL-coated plate(s) in the incubator at 37 °C to warm it before seeding cells. Remove the storage solution of 1x antibiotic-antimycotic from the 384-well PLL-coated plate(s) by flicking the plate over the sink and taping it over a paper towel to dry.
- Gently rinse the transfected cells with Versene solution (1X PBS, pH 7.4; 0.53 mM EDTA), and detach by adding 3 mL of 0.05% trypsin/0.53 mM EDTA to the dish. Transfer the contents to a centrifuge tube containing at least an equal amount of complete DMEM to neutralize the trypsin.
- Spin down the cells at 500 x g for 3 min and resuspend the cells at a density of 0.4 x 106 cells/mL in starving media. Seed cells into the 384-well PLL-coated plate(s) at a final density of 25,000 cells/well by dispensing 45 μL of the cell suspension using an electronic multichannel pipet. Return the plates to the 37 °C for a minimum of 4 h, allowing the cells to properly attach to the wells before proceeding to the stimulation.
7. Secondary screening: Drug plate preparation for 16-point (half log) dose-curve
- In a 96-well plate, add 270 μL of 1X HBSS drug buffer (1x Hank's Balanced Salt Solution [HBSS], 20 mM HEPES pH 7.4, 1x antibiotic-antimycotic), excluding the last row (row H) of the plate, as shown in Figure 4.
NOTE: For peptides, colloidal molecules, and poorly water-soluble compounds, the addition of 0.1–1% BSA is suggested. To prevent drug oxidation, up to 0.01% ascorbic acid can also be added. - From the drug stock, prepare a drug solution (referred to as the "High" concentration) by calculating a final 3x concentration (final concentration of the drug in the cell plate will be 1x). As an example, for a dose–response curve with 10 μM as its highest concentration, prepare the "High" concentration at 30 μM. Pipet 300 μL of "High" concentration into wells in row H.
- In another tube, prepare the "Low" concentration, which represents the "High" concentration divided by 3.16 (half-log). Based on the previous example, the "Low" concentration would be 9.49 μM. Pipet 300 μL of "Low" concentration into wells in row H, adjacent to the "High" wells.
NOTE: The total number of 96-wells needed in row H will depend on the number of cells and stimulation conditions. Four wells (two "High" and two "Low") will have ample drug solution to stimulate an entire 384 well plate. - Perform a serial dilution by pipetting 30 μL of drug solution from the "High" and "Low" wells of row H to the previous row (row G) and mix by manually pipetting up and down, or as recommended, using an electronic multichannel pipette with the "Pipette and Mix" function. Repeat this step up until the first and most diluted row (row A), while discarding tips between dilutions.
NOTE: If desired, the serial dilutions can be stopped before row A, representing an internal control with no drug, in other words, a "true zero". - Using Figure 4 as a reference, stimulate transfected cells by pipetting 20 μL of the "Low" column dilutions from the 96-well plate to rows A–O of the previously seeded 384-well plate, as well as 20 μL of the "High" column dilutions to wells B–P. Incubate the plate at 37 °C for a minimum of 16 h.
8. Secondary screening: Luminescence reading and data analysis
- At 16–24 h following stimulation, decant the transfected cell media by gently flicking the 384-well plate over the sink and taping it over a paper towel. Add 20 μL/well of Glo reagent and incubate the plate at room temperature for 5–20 min. Read the plates using a microplate luminescence counter, with an integration time of 1 s/well.
- Export the saved files from the luminescence counter as a spreadsheet; results will be recorded in relative luminescence units (RLU). Transfer the data of the 384-well plate to a statistics software to analyze the results using its built-in XY analysis for non-linear regression curve fit. Select the built-in 3-parameter dose-response stimulation function "Log(agonist) vs. response (three parameters)",

NOTE: Here Top and Bottom are plateaus in the units of the Y axis, respectively the maximal response and basal level, EC50 is the concentration of the agonist that that generates 50% response between Top and Bottom, and X refers to the log concentration of the agonist. This model assumes that the dose-response curve has a standard Hill slope of 1.
Using the PRESTO-Tango protocol presented herein, a chromaffin granule (CG) extract was screened against 168 non-olfactory GPCR targets, with the majority being orphan receptors. Profiling of said extract was performed by examining β-arrestin2 mobilization at the chosen receptors, based on the principle designed by Barnea et al.18 (Figure 1). Plasmid cDNA of the GPCRs of interest was taken from the PRESTO-Tango GPCR Kit and assembled in two 96 well-plates in the desired layout. In total, four 384-well plates seeded with HTLA cells were transfected, as each half of the 96-well DNA plates was made into a full 384-well plate, resulting in each receptor being transfected in two quadrants (eight 384-wells total). One of the two quadrants was stimulated with the CG extract; put differently, alternating rows C–D, G–H, K–L, and O–P represented the (+) stimulation (Figure 2). Out of the 168 GPCRs that were interrogated in the primary screening, only two receptors were contenders as potential active targets, specifically dopamine receptor D3 (DRD3) and opsin 5 (OPN5). DRD3 produced a significant log2fold change of 4.70, whereas OPN5 produced a slightly lower response of 2.39, both meeting the threshold cut-off of log2 fold change >2. In comparison, the positive control for the primary screen was DRD2 stimulated with quinpirole, a selective agonist of this receptor, and produced a log2 fold change of 4.58 (Figure 3). To reproduce these signal windows and eliminate the possibility of false-positive hits, a secondary screen was conducted with the aforementioned receptors. Besides testing the CG extract, given that DRD3 is a non-orphan receptor, another condition was prepared as a positive control, specifically stimulation with quinpirole, one of its selective agonists. On the other hand, OPN5 is an orphan receptor and as such, no reference agonist can be tested alongside the CG extract as a positive control; only buffer was tested as a negative control. Further pharmacological characterization of these two GPCRs was performed by preparing 16-point dose curves ranging from 10-5 M to 10-12.5 M. Specifically, the CG extract and quinpirole stock solutions at 10 mM were diluted to 30 µM and 9.49 µM, the corresponding "High" and "Low" concentrations in bottom row (row H) of the 96-well drug plate; these formulations will become 10-5 M and 10-5.5 M once 20 µL is dispensed on to the transfected cells in 40 µL of starved medium, for a total of 60 µL within each 384-well. As previously described, serial dilutions were performed such that the most dilute drug formations for 10-12 M and 10-12.5 M are in the top row (row A) (Figure 4). Dose-response curves from the secondary screening were created using GraphPad Prism to evaluate ligand potency and efficacy. In comparison to quinpirole, the CG extract produced similar signal windows and EC50 values, confirming its validity as an active hit at DRD3. However, a flat dose-curve similar to the negative control was produced for OPN5, ruling it out as a possible target for the CG extract (Figure 5).
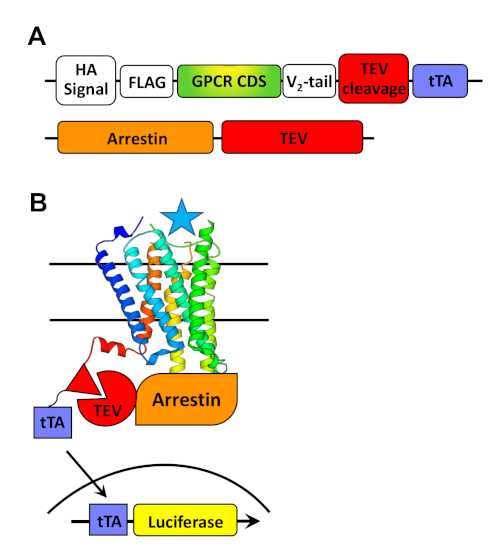
Figure 1: Modular design of TANGO constructs (A) and general scheme for the β-arrestin (Tango) recruitment assay (B). (A) The GPCR Tango constructs consist of various module elements in the following order: an HA signal/FLAG tag, the GPCR CDS, a Vasopressin receptor 2 C-terminal tail, TEV protease cleavage site, and a tTA transcription factor. (B) The principle of the Tango assay involves transiently transfecting the GPCR Tango plasmids in HTLA cells, HEK293T cells stably expressing a β-arrestin2-TEV protease fusion protein and a luciferase reporter gene whose expression is activated by tTA. Activation of the GPCR will eventually result in the mobilization of the β-arrestin2-TEV to the receptor, bringing the protease in close proximity to its cleavage site. As a result, the tTA is cleaved from the GPCR tail, freeing the transcription factor to translocate into the nucleus and activate luciferase expression. Please click here to view a larger version of this figure.
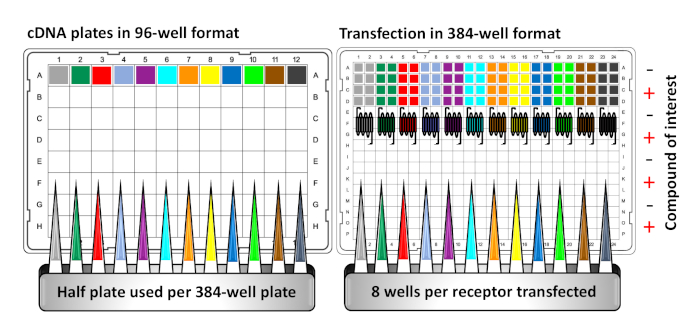
Figure 2: Layouts of 96-well cDNA plate and 384-well cell plate for transfection and stimulation in PRESTO-TANGO primary screening. Depicting the preparation of a 384-well cDNA source plate for transfection, GPCR Tango constructs are first transferred from one half of a 96-well plate into a full 384-well plate, with each receptor being transfected in octuplicate. In this setting, stimulation of cells with (+) and without (-) the drug(s) of interest will occur in quadruplicate for each individual receptor. Please click here to view a larger version of this figure.
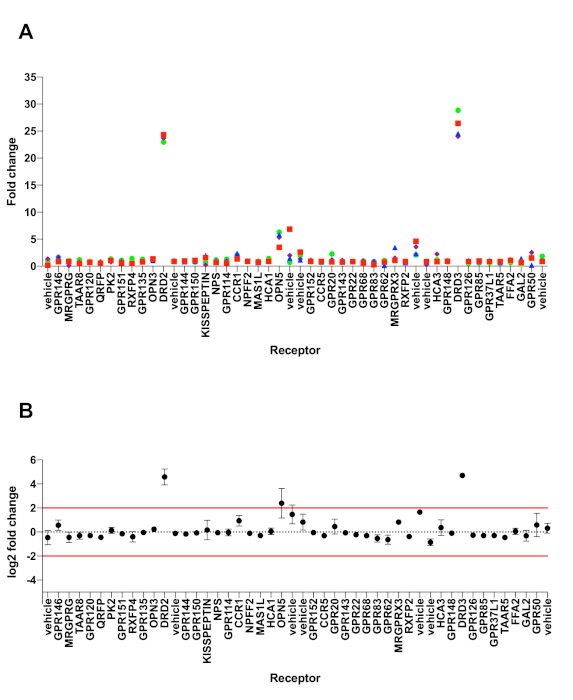
Figure 3: Graphical representations of hit identification from PRESTO-Tango primary screening. As a proof-of-concept, the biological activity of a chromaffin granule (CG) extract on the GPCRome was analyzed. HTLA cells were transfected in 384 well plates with 168 GPCR Tango constructs, and either stimulated with the CG extract (+ compound) or with vehicle buffer (- compound). pcDNA3.1 was used as a negative control, and DRD2 receptor stimulated with quinpirole was used as a positive control. The signal windows (A) and the log2 fold change (B) in receptor activation was calculated between the wells in the absence or presence of CG extract. All error bars represent SD (n = four measurements). Please click here to view a larger version of this figure.
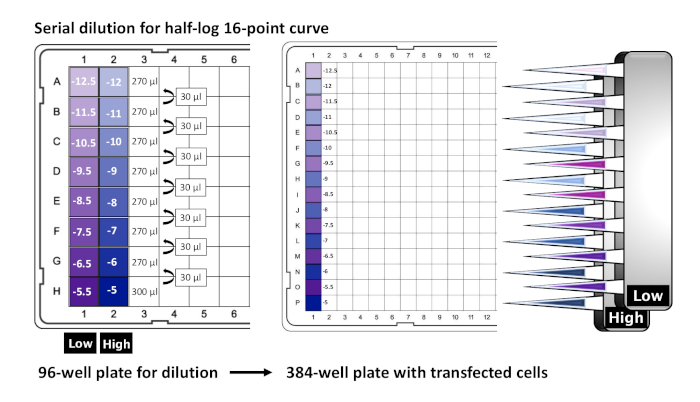
Figure 4: Layout of 96-well drug plate preparation for stimulation in secondary screening. Depicting the preparation of a 96-well drug plate for cell stimulation, serial dilutions for a 16-point dose curve range start at 10-5 M (final concentration) in row H, with half-log intervals between each point until 10-12.5 M in row A. "High" and "Low" drug columns are used to stimulate alternating rows of the seeded 384-well plate. Please click here to view a larger version of this figure.
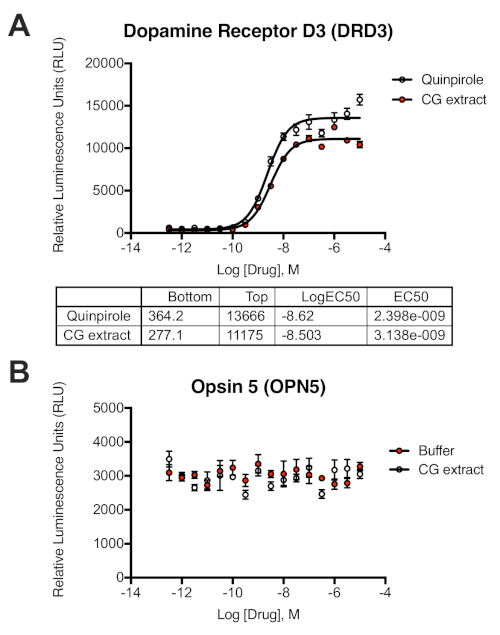
Figure 5: Dose-curve responses for compound profiling and demonstration of β-arrestin2 recruitment to GPCRs in secondary screening. HTLA cells were transiently transfected with receptors DRD3 (A) and OPN5 (B). Both transfected conditions were stimulated with a CG extract in half-log increments, as well as the DRD3 specific agonist quinpirole as a positive control, and vehicle buffer for OPN5 as a negative control. All error bars represent SD (n = three measurements). Please click here to view a larger version of this figure.
