Quantifying Human Norovirus Virus-like Particles Binding to Commensal Bacteria Using Flow Cytometry
Summary
The goal of this protocol is to quantify binding of the eukaryotic pathogen human norovirus to bacteria. After performing an initial virus-bacterium attachment assay, flow cytometry is used to detect virally-bound bacteria within the population.
Abstract
Commensal bacteria are well established to impact infection of eukaryotic viruses. Direct binding between the pathogen and the host microbiome is responsible for altering infection for many of these viruses. Thus, characterizing the nature of virus-bacteria binding is a foundational step needed for elucidating the mechanism(s) by which bacteria alter viral infection. For human norovirus, commensal bacteria enhance B cell infection. The virus directly binds to these bacteria, indicating that this direct interaction is involved in the mechanism of infection enhancement. A variety of techniques can be used to quantify interactions between bacteria and viruses including scintillation counting of radiolabeled viruses and polymerase chain reaction (PCR). Both methods require the use of live virus, which may need to be generated in the laboratory. Currently, none of the established in vitro culture systems available for human norovirus are robust enough to allow for generation of highly concentrated viral stocks. In lieu of live virus, virus-like particles (VLPs) have been used to characterize the interactions between norovirus and bacteria. Herein a flow cytometry method is described with uses virus specific antibodies to quantify VLP binding to gram-negative and gram-positive bacteria. Inclusion of both bacteria only and isotype controls allowed for optimization of the assay to reduce background antibody binding and accurate quantification of VLP attachment to the bacteria tested. High VLP:bacterium ratios result in VLPs binding to large percentages of the bacterial population. However, when VLP quantities are decreased, the percent of bacteria bound also decreases. Ultimately, this method can be employed in future experiments elucidating the specific conditions and structural components that regulate norovirus:bacterial interactions.
Introduction
Human noroviruses (HuNoVs) are the leading cause of gastrointestinal illness worldwide, responsible for 685 million infections and over 200,000 deaths each year1. As with other enteric viruses, the presence of commensal bacteria has been shown to enhance infection of this pathogen as well as its surrogate virus, murine norovirus2,3. There are also conflicting reports that bacteria may inhibit infection by human norovirus4,5,6. For several viruses, direct interaction between the virus and bacteria appear to underlie the mechanisms that impact viral infection2,7,8,9,10, and it has been shown through electron microscopy that human noroviruses bind directly to the surfaces of bacteria11,12. Therefore, characterizing these interactions has become critical to determining the mechanisms by which bacteria impact viral infection. This characterization has classically begun with quantifying viral binding to an array of bacterial species that are components of the host microbiome7,12,13. These attachment assays not only reveal the amount of virus bound to bacteria, but also aid in determining the impact of this interaction on viral fitness and survival.
To quantify viral attachment, traditionally employed methods include PCR-based assays which quantify viral genomes12 or the generation of radiolabeled virus and the use of scintillation counting to quantify viral particles7,8,9,13. The use of these methods generally require access to high-titer virus stocks and in vitro cultivation techniques with which to generate them. While several culture systems for human norovirus now exist2,14,15, none support the robust replication required to generate these highly concentrated stocks which restricts or eliminates the use of PCR and scintillation counting to quantify human norovirus/bacterial interactions.
To circumvent this issue, virus-like particles (VLPs) can be used as a surrogate to live virus to investigate interactions between human norovirus and bacteria16,17. VLPs are non-infectious particles that closely resemble the virus from which they are derived. In the case of human norovirus, these particles are generated from the expression of the VP1 (and sometime the VP2) protein, which self-assemble to create intact viral capsids lacking genetic material (i.e., RNA for noroviruses). These VLPs have been well characterized, are structurally and antigenically similar to the wild-type viruses from which they are derived18,19,20,21,22,23. Therefore, VLPs serve as an ideal surrogate for investigating the surface interactions between human norovirus and commensal bacteria. Given that VLPs lack genetic material, PCR-based assays cannot be used to quantify viral binding. An antibody-based flow cytometry method was previously described and able to detect low levels of VLP binding to bacteria in a semi-quantitative manner16. This method was optimized to allow for accurate quantification of human norovirus VLP binding to both gram-negative and gram-positive commensal bacteria16.
Protocol
NOTE: The bacterial growth conditions outlined in the protocol are standard culture conditions for Enterobacter cloacae and Lactobacillus gasseri. To perform the virus:bacteria attachment assay with other bacterial species, the chosen bacteria should be cultured under standard conditions appropriate for the bacterium.
1. Preparing Bacterial Growth Medium
- Enterobacter cloacae growth media
- Prepare liquid medium by dissolving 10 g of tryptone, 5 g of yeast extract and 10 g of sodium chloride (NaCl) in 1 L of de-ionized (DI) water (see Table of Materials). Mix all media thoroughly and sterilize by autoclaving for 30 min.
- Prepare solid medium by dissolving 10 g of tryptone, 5 g of yeast extract, 10 g of NaCl, and 15 g of agar in 1 L of DI water. Mix all media thoroughly and sterilize by autoclaving for 30 min.
- Lactobacillus gasseri growth media
- Prepare liquid medium by dissolving 55 g of De Man, Rogosa and Sharpe (MRS; see Table of Materials) powder in 1 L of DI water. Use medium within one month of preparation. To sterilize, heat medium for 15 min until boiling followed by autoclaving for 15 min.
- Prepare solid medium by dissolving 55 g of MRS powder and 15 g of agar in 1 L of DI water. To sterilize, heat medium for 15 min until boiling followed by autoclaving for 15 min.
2. Establishing a Standard Curve Correlating Optical Density (OD) and Bacteria Concentration
- Inoculate 5 mL of appropriate liquid medium with a single, isolated colony from an agar plate (E. cloacae) or directly from frozen glycerol stock (L. gasseri).
- Grow the bacterium overnight, under proper atmospheric conditions (Enterobacter cloacae: 37 °C with aerobic shaking at 200 rpm; L. gasseri: 37 °C water bath with no shaking in an airtight container).
- Transfer 2 x 1.3 mL aliquots of overnight culture into two separate 1.5 mL centrifuge tubes and centrifuge bacteria at 10,000 x g for 5 min.
- Remove the supernatant and wash samples with 1 mL of sterile 1x phosphate buffered saline (PBS). Repeat this wash step for a total of 2 washes.
- Centrifuge the samples again at 10,000 x g for 5 min, remove the supernatant and resuspend in 1.3 mL of sterile 1x PBS.
- Beginning with 0.5 mL of washed culture, serially dilute the bacteria in 1x PBS from 10-1 to 10-4.
- Using a spectrophotometer, measure the optical density of the washed, undiluted culture and each of the four dilutions at 600 nm (OD600).
- Perform 10-fold serial dilutions in 1x PBS for each dilution from step 2.6. Spread plate 100 µL of the last 3 dilutions for each series onto appropriate solid medium to determine the number of colony forming units per milliliter (CFU/mL) of each sample. Plate each dilution in triplicate.
- Allow the plates to dry at room temperature for 5 min. Invert the plates and incubate under the appropriate atmospheric conditions (E. cloacae: aerobic at 37 °C, incubate plates overnight; L. gasseri: anaerobic at 37 °C, incubate plates for 48 h in an airtight container with anerobic air sachets (see Table of Materials)).
NOTE: Use 1 anaerobic air sachet per 2.5 L jar or 3 sachets per 7.0 L jar (see Table of Materials). - Use plate counts to determine CFU/mL for each of the 5 samples (i.e., undiluted, 10-1, 10-2, 10-3, 10-4). Generate a standard curve comparing OD600 vs CFU/mL. The equation for this line will be used in VLP-bacteria attachment assays to determine CFU/mL based on OD600.
3. VLP-bacteria Attachment Assay
CAUTION: Human norovirus VLPs are a biosafety level (BSL)-2 hazard and all work involving VLPs should be performed in a biosafety cabinet. Preparation of the bacterial cultures, prior to the attachment assay, should be performed using safety conditions appropriate for the organism.
- Reagent preparation
- 1x phosphate buffered saline (PBS)
- Prepare 1 L of 10x PBS by dissolving an entire packet in 1 L of DI water.
- Autoclave solution for 30 min.
- Prepare a 1x solution by diluting the above solution 1:10 in DI water.
- Filter sterilize the solution by passing it through a 0.22 μm filter and store at room temperature.
- 5% BSA
- Add 5 g of bovine serum albumin (BSA) powder to 100 mL of PBS to generate a 5% (w/v) solution.
- Mix solution by vortexing or on a stir plate using a metal stir bar until clumps have dissolved.
- Filter sterilize using a 0.22 μm filter.
- Store the solution at 4 °C.
- Flow cytometry stain buffer (FCSB)
- Filter purchased FCSB (see Table of Materials) through a 0.22 μm filter.
- Store remaining solution at 4 °C.
- 5% Blocking Buffer (BB)
NOTE: Prepare fresh each time.- Add 0.5 g of BSA to 10 mL of sterile FCSB.
- Vortex to fully mix sample and leave on ice until use.
- 1x phosphate buffered saline (PBS)
- Antibody conjugation
- Conjugate human norovirus GII antibody (see Table of Materials) with r-phycoerythrin (PE) using an R-PE antibody labeling kit per the manufacturer's instructions (see Table of Materials).
- Store the conjugated antibody in the dark at 4 °C for future use in VLP-bacterium attachment assay analysis.
NOTE: The primary antibody should be titrated prior to use in the VLP-bacteria attachment assay to determine the proper concentration necessary for flow cytometry analysis. Antibody titration should be performed every time the conjugation reaction is performed and for each bacterium used in the attachment assay.
- Antibody titration
- Dilute the conjugated anti-human norovirus GII antibody 100-fold, with a starting dilution of 1:100 and an ending dilution of 1:600 (six dilutions in total).
- Perform a virus attachment assay as described below with the following exception: in step 3.5.7 resuspend the bacterial pellet in 350 µL of 5% BB.
- Divide the sample into seven 50 µL aliquots.
- Add 50 µL of each antibody dilution to one tube.
- Add 50 µL of 5% BB to the seventh tube as an unstained control.
- Perform flow cytometry on all samples as described below.
- Compare diluted antibody samples to unstained controls and to each other. The lowest antibody concentration that does not result in a decrease or loss of positive signal should be chosen for use in subsequent assays.
- Preparation of bacteria
- Grow the bacteria in 40 mL of appropriate liquid medium under appropriate atmospheric conditions until the bacteria are in stationary phase.
- Grow E. cloacae cultures aerobically overnight in Luria Broth at 37 °C, shaking at 200 rpm to reach stationary phase.
- Grow L. gasseri cultures in MRS broth in black screw cap tubes without shaking in a water bath set to 37 °C for 18 h to reach stationary phase.
- Transfer stationary phase cultures to clean 50 mL conical tubes and centrifuge samples at 2,288 x g for 10 min to pellet the bacteria.
- Remove the supernatant and resuspend in 13 mL of sterile 1x PBS. Repeat this wash step for a total of two washes.
- Centrifuge the samples again, remove the supernatant and resuspend samples in 20 mL of 1x PBS.
NOTE: If the cell pellet is small, the resuspension volume can be decreased. - Measure the OD600 of the culture.
- Using the previously prepared standard curve, determine the CFU per mL of the washed culture.
- Dilute the bacteria as needed with 1x PBS to adjust culture concentration to 1 x 108 CFU/mL.
- Transfer 1 mL of the 1 x 108 CFU/mL bacterial culture to the required number of 1.5 mL centrifuge tubes.
- Centrifuge the tubes at 10,000 x g for 5 min.
- Resuspend the bacterial pellet in 1 mL of sterile 5% BSA.
- Incubate the tubes for 1 h at 37 °C with constant rotation.
- Grow the bacteria in 40 mL of appropriate liquid medium under appropriate atmospheric conditions until the bacteria are in stationary phase.
- Virus attachment
- Working inside a BSL-2 biosafety cabinet, add 10 µg of HuNoV VLPs (see Table of Materials) to each tube containing bacteria resuspended in PBS and mix thoroughly by pipetting.
NOTE: VLP concentration differs with each VLP preparation. Therefore, the volume added to bacteria will vary between preparation batches and should be adjusted accordingly so that 10 µg is added to each tube in the experiment. For bacteria only controls (e.g., samples without VLP), add a volume of PBS that equals the volume of VLP added to experimental samples. - Incubate tubes for 1 h at 37 °C with constant rotation.
NOTE: A tube revolver (see Table of Materials) set to 40 rpm is used during incubation. - After incubation, centrifuge the samples at 10,000 x g for 5 min.
- Discard supernatant and resuspend bacterial pellet in 1 mL of PBS.
- Repeat the wash steps 3.5.3 and 3.5.4.
- Centrifuge the samples at 10,000 x g for 5 min.
- Discard supernatant and resuspend bacterial pellet in 150 µL of 5% BB.
- Working inside a BSL-2 biosafety cabinet, add 10 µg of HuNoV VLPs (see Table of Materials) to each tube containing bacteria resuspended in PBS and mix thoroughly by pipetting.
- Antibody staining
- Prepare fresh 5% BB.
NOTE: All work using the fluorescently tagged antibody should be performed in the dark and the antibody, BB and FCSB should be kept on ice. - Dilute human norovirus GII antibody 1:125 for E. cloacae samples and 1:150 for L. gasseri samples in 5% BB, preparing 50 µL of diluted antibody per sample.
- Dilute the isotype antibody in the same way the human norovirus GII antibody was diluted for each bacterium in 5% BB, preparing 50 µL of diluted isotype control per sample.
- Divide each attachment assay sample from step 3.3.7 into three 50 µL aliquots by transferring them into clean 1.5 mL centrifuge tubes.
- To the first set of samples, add 50 µL of BB. This set will be the unstained (Uns) controls.
- To the second set, add 50 µL of the GII antibody dilution. This results in a final antibody concentration of 1:250 for E. cloacae and 1:300 for L. gasseri. This sample set will be the stained (AB) samples.
- To the third set, add 50 µL of the diluted isotype antibody. This set will be the isotype controls (IC). Mix well by pipetting. Incubate the samples on ice and in the dark for 30 min.
- Centrifuge all samples at 10,000 x g for 5 min.
- Discard the supernatant and resuspend the samples in 100 µL of FCSB.
- Centrifuge all samples at 10,000 x g for 5 min. Discard the supernatant and resuspend the samples in 100 µL of FCSB.
- Centrifuge all samples at 10,000 x g for 5 min.
- Discard the supernatant and resuspend the samples in 150 µL of FCSB.
- Transfer each sample to a FCSB tube containing 400 µL of FCSB buffer for a total volume of 550 µL.
- Keep samples at 4 °C until they are analyzed by flow cytometry.
NOTE: Flow cytometry is performed within 4 h of antibody staining.
- Prepare fresh 5% BB.
4. Flow Cytometry
NOTE: The voltage settings described below are based on the flow cytometer and software listed in the Table of Materials and will likely vary with different flow cytometers. Settings should be optimized for each bacterium. Ensure that the axes for all graphs are in biexponential phase.
- Setting up the workspace
NOTE: The set-up for plots used to establish the workspace differ from those used in gating and data analysis. The purpose of setting up the workspace is to visualize the cell population, ensure there is no excessive clumping of the bacterial cells that might impact downstream analysis and to distinguish between stained and unstained cells while data is being collected.- In order to ensure bacteria are not clumping together, set up a series of density plots.
- Generate a forward scatter area (FSC-A) versus side scatter area (SSC-A) to visualize the total population.
- Set up plots to evaluate single cells versus cell clumps by creating two graphs that compare both forward (FSC-W) and side scatter width (SSC-W) to forward (FSC-H) and side scatter height (SSC-H), respectively.
- Create a plot that shows only the PE positive population (PE-A vs. SSC-A).
- Set baseline voltage to 500 for E. cloacae and 340 for L. gasseri. These will be further adjusted later.
- Set the total events counted to 10,000 events.
- In order to ensure bacteria are not clumping together, set up a series of density plots.
- Running samples
- Create three separate histogram plots that show count plotted against SSC-A, FSC-A or PE-A.
- Place an unstained sample on the flow cytometer and acquire the sample events, ensuring that the maximum peak on the histogram for SSC-A is within 102 to 103 and the maximum peak on the histogram for FSC-A is between 104 to 105 on the x-axis. If not, peaks do not fall within the specified ranges, adjust the FSC and SSC voltages accordingly.
- Place a stained sample (AB) on the flow cytometer and adjust the voltage PE to make the maximum peak past 103 on the x-axis.
- Based on these measurements, set the positive PE gate and set the gate on the PE-A versus SSC-A density graph.
- Run all samples under the determined settings at low or medium speed.
Representative Results
The gating strategies used to quantify human norovirus VLP binding to commensal bacteria are shown in Figure 1. Representative density dot provides an overview of how samples were gated to eliminate cellular debris and cell clumps so VLP attachment was determined on singlet populations (Figure 1A). Representative histograms demonstrate low levels of anti-norovirus antibody signal in bacteria only samples lacking norovirus VLP and low background signal of VLP-bacteria samples stained with the isotype control antibody (Figure 1B). Isotype control peaks also overlap with unstained samples while staining of the same samples with anti-human norovirus GII antibody results in significant shift in peak.
The Overton method of histogram subtraction was used to compare the PE-positive signal in anti-human norovirus GII antibody stained samples to the PE-positive signal of the corresponding isotype control and determine the percent of the bacterial population bound by human norovirus VLPs (Figure 1B). After 1 h of incubation with 10 µg of VLP, flow cytometry detected particle binding to both E. cloacae and L. gasseri (Figure 2). In fact, high levels of binding occurred under these conditions for both bacteria; binding to L. gasseri occurred at slightly higher, but significant (p < 0.0001), levels compared to E. cloacae. These assays demonstrate that flow cytometry can be used to detect human norovirus binding to both Gram-positive and Gram-negative bacteria.
To determine the limit of binding quantification for this assay, a dilution series of the VLPs was generated prior to addition to the bacteria (Figure 3). For both genera of bacteria, reductions in the amount of VLP added to the bacterial culture resulted in corresponding reductions in the percent of bacteria bound by VLP. Changes in the percent of E. cloacae bound by the particle were more gradual compared to L. gasseri, but percent attachment for both bacteria leveled off despite further reductions in VLP concentration. Specifically, 0.1 µg or less (data not shown) of VLP in 108 CFU of bacteria resulted in a plateau of percent attachment averaging between 13–19% for both bacteria, indicating that this percentage is the limit of quantification for this assay.
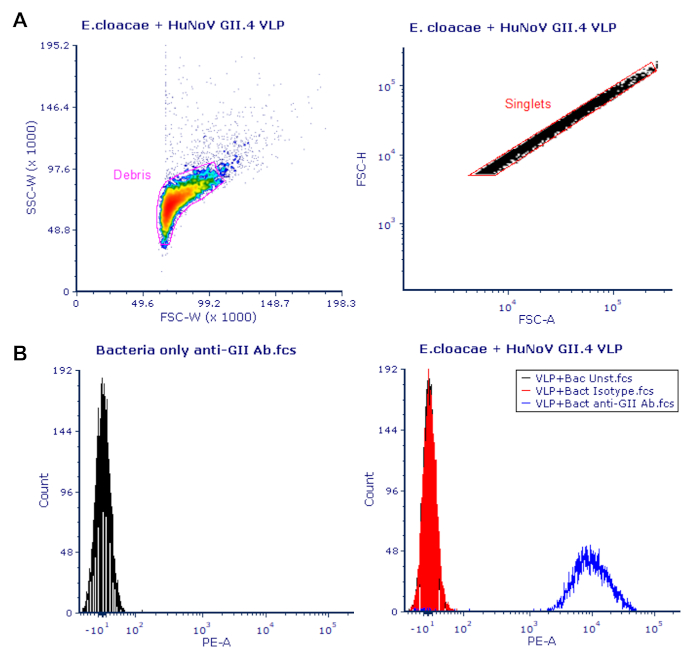
Figure 1: Representative flow cytometric analysis of human norovirus VLP binding. (A) Representative flow cytometry gating strategy to quantify VLP binding to bacteria. Density plots were used to gate out cellular debris, followed by subsequent dot plot gating to remove bacterial doublets and cellular clumps. (B) Representative histograms demonstrate a lack of PE signal in bacterial only samples and a shift in PE signal intensity in VLP:bacterium samples compared to unstained and isotype controls. Please click here to view a larger version of this figure.
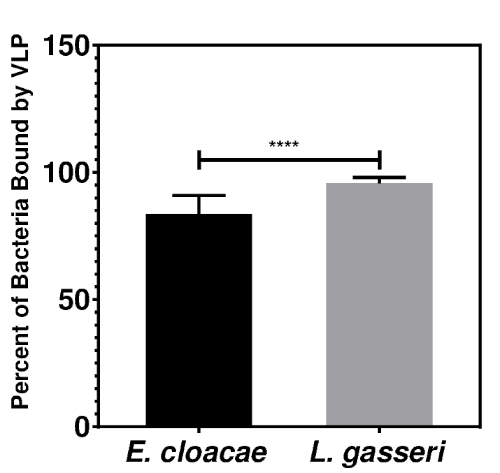
Figure 2: VLP attachment to E. cloacae and L. gasseri. Overnight cultures of E. cloacae (n = 6) and L. gasseri (n = 6) were diluted to 1 x 108 CFU/mL in PBS. The bacteria were incubated with 10 µg of GII.4 VLPs for 1 h then VLP attachment was measured using flow cytometry. A representative plot can be found in Figure 1B. Percent attachment was determined using the Overton method of histogram subtraction comparing the PE-positive signal of the GII Human Norovirus stained samples to the PE-positive signal of the corresponding isotype control. Statistical analysis was performed using an unpaired Student's t-test (p < 0.0001). Please click here to view a larger version of this figure.
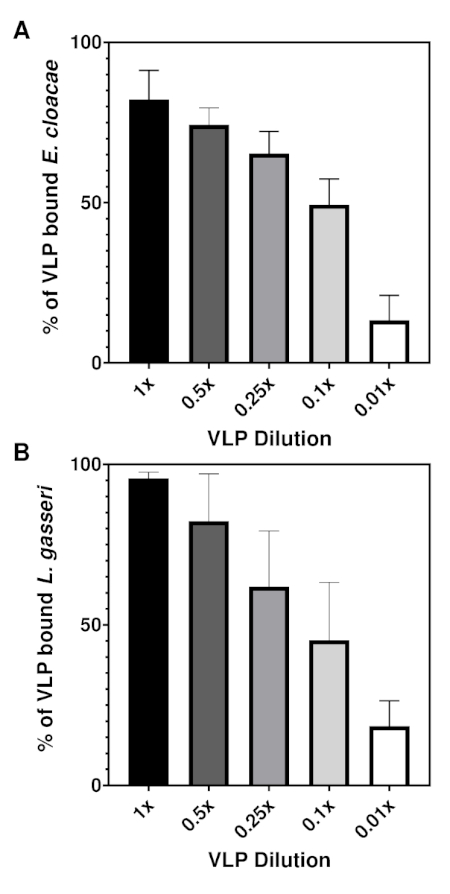
Figure 3: VLP dilution series for E. cloacae and L. gasseri. 10 µg of GII.4 VLPs was serially diluted and the VLP dilutions were each added to 1 x 108 bacteria in a final volume of 1 mL (n = 3 for both bacteria). Samples were incubated for one hour at 37 °C. VLP attachment to the bacteria was measured using flow cytometry. Percent attachment was determined using the Overton method of histogram subtraction comparing the PE-positive signal of the GII Human Norovirus stained samples to the PE-positive signal of the corresponding isotype control. Reducing input amounts of VLP resulted in stepwise reductions in percent attachment for both (A) E. cloacae and (B) L. gasseri. Please click here to view a larger version of this figure.
Discussion
The ability to quantify binding of enteric viruses to bacteria is a critical first step for elucidating the mechanisms by which these bacteria alter viral infection. The methods described herein have been optimized to measure human norovirus VLP interactions with both E. cloacae (gram-negative bacterium) and L. gasseri (a gram-positive bacterium), but can be adapted for use with any mammalian virus and bacterium of interest. While VLPs are an ideal alternative to live virus for use in attachment assays and these particles can be readily quantified using flow cytometry, P particles have also been used to examine interactions between human noroviruses and bacteria24. In this study, the amount of virus bound to the bacteria was quantified as opposed to the bacterial population, as is reported here. P particles provide an advantage over VLPs in that they are easier to produce while providing antigenic similarity to both VLPs and wild-type virus24,25. However, human norovirus VLPs are commercially available providing a means for laboratories lacking the capacity to generate VLPs or P particles. P particles differ from VLPs in that, while VLPs maintain the size of a wild-type viral particle, P particles are smaller and have tetrahedral rather than icosahedral symmetry25. The impact of these characteristics on interactions with bacteria have not been explored and P particles can serve as a viable alternative to VLPs in characterizing surface interactions between human norovirus and bacteria.
As mentioned previously, the assay described above can be used to further characterize interactions between human norovirus VLPs and bacteria. Investigations into how growth conditions and changes in bacterial surface structure expression alter viral binding can be explored using this technique. In addition, this assay can also be used to determine specific bacterial structures bound by the virus through competitive inhibition assays using bacterial proteins or glycans, enzymatic treatment to remove specific surface structures, or incubation with mutant bacterial strains deficient in particular a structure. This assay can also be employed to investigate the ability of other norovirus strains to interact with commensal bacteria.
Because this assay quantifies the proportion of the bacterial population bound by norovirus VLP, it is critical to accurately determine the correlation between CFU/mL and OD600 so bacterial culture concentration can be measured. Fluctuations in the VLP:bacteria ratio alters the percent of the bacteria population bound by VLPs and can lead to variability in results. Care should also be taken to add sufficient quantities of viral particles, as the limit of detection of this assay approaches 0.1 µg of VLP/108 CFU of bacteria. Ratios below this limit consistently yielded percent attachment values of 13–19%; thus observed population attachment at or below these percentages may not be real.
Antibody titrations were performed for each newly conjugated antibody and against each bacterial strain prior to use in VLP:bacteria attachment experiments. Antibody concentrations required for each bacterium were similar ranging from 1:250 for E. cloacae and 1:300 for L. gasseri. The small size of bacteria, relative to the size of eukaryotic cells, requires both voltage adjustment as well as the use of a binomial distribution during data collection to adequately separate bacteria from debris that may be found in samples or circulating within the instrument. After data collection, proper gating can be used to further remove larger debris particles and bacterial clumps so only single cell populations are analyzed. It is also critical to establish unique voltage settings for each bacterial species tested as these fluctuate widely, particularly between gram-negative and gram-positive bacteria.
Inclusion of proper controls including bacteria only and isotype controls are also critical for accurate analysis of the data. Both types of controls inform regarding levels of non-specific antibody binding. Our results demonstrate that the antibodies used to do not bind non-specifically to the bacterial species tested, but non-specific binding could change with changes in bacterial species or antibody.
The method presented here quantifies human norovirus particle binding to both gram-positive and gram-negative bacteria and is useful in characterizing virus:bacterial interactions. Furthermore, this base protocol can be easily optimized for use with other genotypes of human norovirus, as well as other mammalian viruses and bacteria.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to thank Sutonuka Bhar and Chanel Mosby-Haundrup for their critical review of the written manuscript, as well as Alfonso Carrillo for assistance with generating bacterial standard curves. This work is funded in part by a grant from the National Institute of Health (R21AI140012) and by a seed grant from the University of Florida, Institute of Food and Agricultural Sciences.
Materials
| 5ml Polystrene Round-Bottom Tubes with Cell-Strainer Cap | Corning | 352235 | After antibody staining, sample are transferred into tubes for flow cytometry analysis. |
| Agar | Sigma | A7002 | Used for media preparation |
| AnaeroPack | Thermo Scientific | R681001 | Anaerobic gas pack used for culture of Lactobacillus gasseri |
| BD FacsDiva software | |||
| BD LSR Fortessa flow cytometer | |||
| Bovine Serum Albumin | Fisher Bioreagents | BP1605 | Used for flow cytometry |
| Flow Cytometry Stain Buffer (FCSB) | BD Biosciences | 554657 | Used for flow cytometry |
| Mouse IgG2b kappa Isotype Control (eBMG2b), PE, eBioscience | Thermo Fisher Scientific | 12473281 | Isotype control. This antibody is purchased in the conjugated form from the manufacturer. |
| MRS Powder | BD Biosciences | 288130 | Used for media preparation and to culture Lactobacillus gasseri. |
| Norovirus capsid G2 Monoclonal Antibody (L34D) | Thermo Fisher Scientific | MA5-18241 | Norovirus GII antibody. This antibody is only available in the unconjugated form and thus must be fluorescently conjugated prior to use in the outlined flow cytometry assays. In this protocol, PE was the chosen fluor, however, other fluorescent molecules can be chosen as best suits the flow cytometer being used by the researcher. |
| Norovirus GII.4 VLP | Creative Biostructure | CBS-V700 | human norovirus virus like particle, VLPs were generated using the baculovirus system and resuspended in phosphate buffered saline with 10% glycerol. The authors performed independent nanosight tracking analysis to determine the particle concentration of the VLPs. The concentration is approximately 1011 VLPs per milliliter. Based on the protein concentration of the VLPs, approximately 200 particles are added per bacterium in VLP attachment assays. |
| PBS 10X | Fisher Bioreagents | BP665 | Dilute to 1X prior to use. |
| SiteClick R-PE Antibody Labeling Kit | Thermo Fisher Scientific | S10467 | Conjugation kit used for labling of unconjugated antibody. |
| Sodium Chloride | Fisher Scientific | S271 | Used for media preparation |
| Tryptone | Oxoid | LP0042 | Used for media preparation |
| Tube Revolver | ThermoFisher Scientific | 88881001 | Used in virus:bacterium attachment assay. Set to max speed (40 rpm). |
| Yeast Extract | BD Biosciences | 212750 | Used for media preparation |
References
- Hall, A. J., Glass, R. I., Parashar, U. D. New insights into the global burden of noroviruses and opportunities for prevention. Expert Review of Vaccines. 15 (8), 949-951 (2016).
- Jones, M. K., et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science. 346 (6210), 755-759 (2014).
- Baldridge, M. T., et al. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science. 347 (6219), 266-269 (2015).
- Lei, S., et al. Enterobacter cloacae inhibits human norovirus infectivity in gnotobiotic pigs. Scientific reports. 6, 25017 (2016).
- Lei, S., et al. High Protective Efficacy of Probiotics and Rice Bran against Human Norovirus Infection and Diarrhea in Gnotobiotic Pigs. Frontiers in Microbiology. 7, 1699 (2016).
- Rodríguez-Díaz, J., et al. Relevance of secretor status genotype and microbiota composition in susceptibility to rotavirus and norovirus infections in humans. Scientific Reports. 7, 45559 (2017).
- Erickson, A. K., et al. Bacteria Facilitate Enteric Virus Co-infection of Mammalian Cells and Promote Genetic Recombination. Cell Host Microbe. 23 (1), 77-88 (2018).
- Kuss, S. K., et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 334 (6053), 249-252 (2011).
- Robinson, C. M., Jesudhasan, P. R., Pfeiffer, J. K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe. 15 (1), 36-46 (2014).
- Berger, A. K., Yi, H., Kearns, D. B., Mainou, B. A. Bacteria and bacterial envelope components enhance mammalian reovirus thermostability. PLOS Pathogens. 13 (12), 1006768 (2017).
- Miura, T., et al. Histo-blood group antigen-like substances of human enteric bacteria as specific adsorbents for human noroviruses. Journal of Virology. 87 (17), 9441-9451 (2013).
- Almand, E. A., Moore, M. D., Outlaw, J., Jaykus, L. A. Human norovirus binding to select bacteria representative of the human gut microbiota. PLOS One. 12 (3), (2017).
- Robinson, C. M., Woods Acevedo, M. A., McCune, B. T., Pfeiffer, J. K. Related enteric viruses have different requirements for host microbiota in mice. Journal of Virology. , (2019).
- Ettayebi, K., et al. Replication of human noroviruses in stem cell-derived human enteroids. Science. 353 (6306), 1387-1393 (2016).
- Van Dycke, J., et al. A robust human norovirus replication model in zebrafish larvae. PLOS Pathogens. 15 (9), 1008009 (2019).
- Li, D., Breiman, A., le Pendu, J., Uyttendaele, M. Binding to histo-blood group antigen-expressing bacteria protects human norovirus from acute heat stress. Frontiers in Microbiology. 6, 659 (2015).
- Almand, E. A., Moore, M. D., Jaykus, L. -. A. Characterization of human norovirus binding to gut-associated bacterial ligands. BMC Research Notes. 12 (1), 607 (2019).
- Debbink, K., et al. Within-host evolution results in antigenically distinct GII.4 noroviruses. Journal of Virology. 88 (13), 7244-7255 (2014).
- Harrington, P. R., Lindesmith, L., Yount, B., Moe, C. L., Baric, R. S. Binding of Norwalk virus-like particles to ABH histo-blood group antigens is blocked by antisera from infected human volunteers or experimentally vaccinated mice. Journal of Virology. 76 (23), 12335-12343 (2002).
- Harrington, P. R., Vinje, J., Moe, C. L., Baric, R. S. Norovirus capture with histo-blood group antigens reveals novel virus-ligand interactions. Journal of Virology. 78 (6), 3035-3045 (2004).
- Mallory, M. L., Lindesmith, L. C., Graham, R. L., Baric, R. S. GII.4 Human Norovirus: Surveying the Antigenic Landscape. Viruses. 11 (2), (2019).
- Prasad, B. V., et al. X-ray crystallographic structure of the Norwalk virus capsid. Science. 286 (5438), 287-290 (1999).
- Baric, R. S., et al. Expression and self-assembly of norwalk virus capsid protein from venezuelan equine encephalitis virus replicons. Journal of Virology. 76 (6), 3023-3030 (2002).
- Rubio-del-Campo, A., et al. Noroviral p-particles as an in vitro model to assess the interactions of noroviruses with probiotics. PLOS One. 9 (2), 89586 (2014).
- Tan, M., et al. Terminal modifications of norovirus P domain resulted in a new type of subviral particles, the small P particles. Virology. 410 (2), 345-352 (2011).
