1. Buffers and media
- For MAPS experiments, prepare the following buffers and media:
– Buffer A (150 mM KCl, 20 mM Tris-HCl pH 8, 1 mM MgCl2 and 1 mM DTT)
– Buffer E (250 mM KCl, 20 mM Tris-HCl pH 8, 12 mM maltose, 0.1% Triton, 1 mM MgCl2 and 1 mM DTT)
– RNA loading buffer (0.025% xylene cyanol and 0.025% bromophenol blue in 8 M urea)
– Brain Heart Infusion (BHI) medium (12.5 g of calf brain, 10 g of peptone, 5 g of beef heart, 5 g of NaCl, 2.5 g of Na2HPO4 and 2 g of glucose for 1 L)
– Lysogeny Broth (LB) medium (10 g of peptone, 5 g of yeast extract and 10 g of NaCl for 1 L) - For Northern blot assays, prepare the following buffers:
– Blocking solution (1x maleic acid and 1% blocking reagent)
– Hybridization solution (50% formamide, 5x SSC, 7% SDS, 1% blocking solution and 0.2% N-lauryl sarcosine, 50 mM sodium phosphate). Heat with agitation to dissolve.
CAUTION: Carefully follow the safety precautions related to each product.
– 1 M sodium phosphate (58 mM sodium phosphate dibasic and 42 mM sodium phosphate monobasic)
– Saline, sodium citrate (SSC) buffer, 20x concentrate (3 M NaCl and 300 mM trisodium citrate)
2. Safety issues
- Carry out all steps involving viable pathogenic bacteria in a level 2 containment lab.
NOTE: Only cell extracts can be taken outside after lysis (step 5). - Put on a lab coat and gloves.
- Ensure that the wrists are covered.
- Clean the biological safety cabinet (Class II) with a disinfectant solution.
- Dispose solid wastes exposed to bacteria in the appropriate biomedical bin.
- Treat flasks containing contaminated liquids with a disinfectant solution. Then, discard it in a sink.
- Carefully wash hands and wrists with soap and remove the lab coat before leaving the level 2 containment lab.
3. Plasmid construction
NOTE: For cloning purposes, it is crucial to first identify the boundaries of the endogenous sRNA. The pCN51-P3 and pCN51-P3-MS2 plasmids are described in Tomasini et al. (2017)27. The P3 promoter allows high expression of the sRNA in a cell-density-dependent manner (i.e., when bacteria enter the stationary phase of growth). Many staphylococcal sRNAs accumulate during this growth phase.
- Amplify the sRNA sequence by PCR using a high-fidelity DNA polymerase and a PCR machine. Carefully follow manufacturer’s instructions and read Garibyan and Avashia (2013)31 for more details.
- Use the following templates to design the specific primers:

and 5’-CGCGGATCC(N)-3’ for forward and reverse primers, respectively.
NOTE: These oligonucleotides enable to fuse the MS2 sequence (in bold) to the 5’ end of the sRNA of interest. PstI and BamHI restriction sites (underlined) are added at the 5’ and 3’ extremities of the MS2-sRNA construct to clone the amplicon into the pCN51-P3 plasmid27. (N) corresponds to the gene-specific sequence (15-20 nucleotides). - Digest 1 µg of pCN51-P3 plasmid and 1 µg of the MS2-sRNA PCR product with 2 U of PstI and 1 U of BamHI in the appropriate buffer according to manufacturer’s recommendations.
- Incubate 1 h at 37 °C and purify DNA using a PCR purification kit (see Table of Materials).
- Mix the digested pCN51-P3 plasmid (300 ng) and MS2-sRNA amplicon (molar ratio for vector:insert = 1:3) in a 1.5 mL tube. See Revie et al. (1988)32 to maximize ligation efficiency. Add 1 µL of the Ligase Buffer and 10 U of T4 Ligase in each tube. Adjust the volume to 10 µL with ultrapure water.
- Incubate at 22 °C for at least 2 h.
- Add 5 µL of ligation mixture to 50 µL of frozen DH5α chemically competent E. coli cells. Read Seidman et al. (2001)33 to learn more about plasmid transformation and chemically competent cells.
- Incubate 30 min on ice.
- Heat shock (45 s at 42 °C) the transformation tube using a heat block or water bath.
- Add 900 µL of LB medium and incubate at 37 °C for 30 min.
- Plate 100 µL of the bacterial suspension on a LB agar plate supplemented with ampicillin (100 µg/µL).
NOTE: the pCN51-P3 vector encodes an ampicillin resistance gene, which enables to select only E. coli clones carrying the pCN51-P3-MS2-sRNA plasmid. - Extract the pCN51-P3-MS2-sRNA plasmid from an overnight bacterial culture (5 mL) grown in the presence of ampicillin (100 µg/µL) using a plasmid DNA miniprep kit (see Table of Materials).
- Verify the construct by Sanger sequencing34 using the following primer, 5’-TCTCGAAAATAATAGAGGG-3’.
- Transform the pCN51-P3-MS2-sRNA plasmid into DC10B chemically competent E. coli cells. Repeat steps 3.7 to 3.11.
- Extract the pCN51-P3-MS2-sRNA plasmid (see step 3.12) and transform 1-5 µg of plasmid DNA into HG001 ΔsRNA electrocompetent S. aureus cells using an electroporation apparatus. Carefully follow manufacturer’s instructions. Read Grosser and Richardson (2016)35 to learn more about methods for preparing electrocompetent S. aureus.
CAUTION: This step involves handling of pathogenic bacteria (see step 2). - Add 900 µL of BHI medium and incubate at 37 °C for 3 h.
- Centrifuge 1 min at 16,000 x g. Discard the supernatant.
- Resuspend the pellet in 100 µL of BHI and plate the bacterial suspension on BHI agar plates supplemented with erythromycin (10 µg/µL).
NOTE: The pCN51-P3 vector also encodes an erythromycin resistance gene, which enables to select only S. aureus clones carrying the pCN51-P3-MS2-sRNA plasmid.
4. Bacteria harvesting
CAUTION: This step involves handling of pathogenic bacteria (see step 2).
- Grow one colony of strains carrying either pCN51-P3-MS2-sRNA or pCN51-P3-MS227 plasmids in 3 mL of BHI medium supplemented with erythromycin (10 µg/µL) in duplicates.
- Dilute each overnight culture in 50 mL (≈1/100) of fresh BHI medium supplemented with erythromycin (10 µg/µL) to reach an OD600nm of 0.05. Use 250 mL sterilized flasks (5:1 flask-to-medium ratio).
NOTE: Medium and growth conditions should be set according to the expression pattern of the studied sRNA. - Grow cultures at 37 °C with shaking at 180 rpm for 6 h.
- Transfer each culture into a 50 mL centrifuge tube.
- Centrifuge at 2,900 x g during 15 min at 4 °C. Discard the supernatant.
- Keep pellets on ice and directly perform mechanical cell lysis (step 5) or freeze and store pellets at -80 °C.
5. Mechanical cell lysis
CAUTION: Following steps must be performed on ice and buffers must be at 4 °C. Use gloves and take all precautions to protect samples from RNases.
- Resuspend pellets (step 4.6) in 5 mL of Buffer A.
- Transfer the resuspended cells in 15 mL centrifuge tubes with 3.5 g of silica beads (0.1 mm).
- Insert tubes in a mechanical cell lysis instrument (see Table of Materials). Run a cycle of 40 s at 4.0 m/s.
NOTE: If one cycle is not enough to break cells, let the device cool for 5 min while keeping samples on ice. Then, repeat another cycle of 40 s at 4.0 m/s. The efficiency of cell lysis can be tested by plating the supernatant on BHI-agar plate. - Centrifuge at 15,700 x g for 15 min. Recover the supernatant and keep it on ice.
6. Column preparation
CAUTION: Be careful not to allow the amylose resin to dry. If needed, seal the column with an end-cap. Prepare all the solutions before starting the affinity purification.
- Put a chromatography column in a column rack (see Table of Materials).
- Remove the column tip and wash the column with ultrapure water.
- Add 300 µL of amylose resin.
- Wash the column with 10 mL of Buffer A.
- Dilute 1,200 pmol of MBP-MS2 protein in 6 mL of Buffer A and load it into the column.
- Wash the column with 10 mL of Buffer A.
7. MS2-affinity purification (Figure 1)
- Load the cell lysate into the column.
NOTE: Keep 1 mL of the cell lysate (Crude extract, CE) to extract total RNA (step 8) and perform Northern blot (step 9) and transcriptomic (step 10) analysis. - Collect the flow-through fraction (FT) in a clean collection tube.
- Wash the column 3 times with 10 mL of Buffer A. Collect the wash fraction (W).
- Elute the column with 1 mL of Buffer E and collect the elution fraction (E) in a 2 mL microtube.
- Keep all collected fractions on ice until RNA extraction (step 8) or freeze them at -20 °C for later use.
8. RNA extraction of collected fractions (CE, FT, W and E)
- Use 1 mL of each fraction (including FT and W) for RNA extraction.
- Add 1 volume of phenol. Mix vigorously.
CAUTION: Phenol is volatile and corrosive, pay attention and work safely under a fume hood. - Centrifuge at 16,000 x g for 10 min at 20 °C.
- Transfer the upper phase in a clean 2 mL microtube.
- Add 1 volume of chloroform/isoamyl alcohol (24:1) and repeat steps 8.3 to 8.4.
CAUTION: Work safely under a fume hood. - Add 2.5 volumes of cold ethanol 100% and 1/10 volume of 3 M sodium acetate (NaOAc) pH 5,2.
- Precipitate overnight at -20 °C.
NOTE: Precipitation can also be performed in an ethanol/dry ice bath during 20 min or at -80 °C during 2 h. - Centrifuge at 16,000 x g for 15 min at 4 °C. Slowly remove ethanol with a pipette while being careful not to disturb the pellet.
CAUTION: The RNA pellet is not always visible and is sometimes loose in presence of ethanol. - Add 500 µL of 80% cold ethanol.
- Centrifuge at 16,000 x g for 5 min at 4 °C.
- Discard ethanol by pipetting it slowly. Dry the pellet using a vacuum concentrator, 5 min on run mode.
- Resuspend the pellet in an appropriate volume (15-50 µL) of ultrapure water. Freeze the pellet at -20 °C for later use.
- Assess RNA quantity (260 nm) and quality (260/280 and 260/230 wavelength ratios) using a spectrophotometer/fluorometer (see Table of Materials). Carefully follow manufacturer’s instructions.
NOTE: 3-4 µg are generally obtained in the elution fraction (E). This mainly depends on tested conditions.
9. Analysis of MS2-affinity purification by Northern blot36
- Dilute 5 µg of RNA of CE, FT, W fractions and 500 ng of E fraction in 10 µL of ultrapure water and mix with 10 µL of RNA loading buffer.
- Incubate 3 min at 90 °C.
- Load samples into wells of an 1% agarose gel supplemented with 20 mM of guanidium thiocyanate and run the gel at 100-150 V in TBE 1x buffer at 4 °C. Read Koontz (2013)37 for more details.
- Transfer RNAs on a nitrocellulose membrane by vacuum transfer for 1h or capillarity transfer overnight.
NOTE: The capillarity method is more efficient for large RNAs. - UV cross-link RNAs on the membrane (120 mJ at 254 nm) using an ultraviolet crosslinker.
- Insert the membrane in a hybridization bottle with the RNA side facing up.
- Add 10-20 mL of preheated hybridization solution. Incubate 30 min at 68 °C.
- Discard the solution and add 10-20 mL of fresh hybridization solution supplemented with 1 µL of the sRNA-specific probe. Incubate overnight at 68 °C.
NOTE: The DIG-labelled RNA probe is synthetized using a DIG RNA labelling kit and following manufacturer’s instructions. Alternatively, a radiolabelled probe can be used. - Wash the membrane with 10-20 mL of wash solution 1 (2x SSC and 0.1% SDS) for 5 min at 20 °C. Repeat once.
- Wash the membrane with 10-20 mL of wash solution 2 (0.2x SSC and 0.1% SDS) for 15 min at 68 °C. Repeat once.
- Incubate with 10-20 mL of blocking solution for at least 30 min at 20 °C.
- Discard the solution and add 10-20 mL of the blocking solution supplemented with the polyclonal anti-digoxigenin antibody (1/1000), conjugated to alkaline phosphatase. Incubate 30 min at 20 °C.
- Wash the membrane with 10-20 mL of the wash solution 3 (1x maleic acid and 0.3% Tween 20) for 15 min at 20 °C. Repeat once.
- Incubate the membrane with 10-20 mL of the detection solution (0.1 M Tris HCl and 0.1 M NaCl pH 9.5) 5 min at 20 °C.
- Put the membrane on a plastic film and soak it with the substrate (see Table of Materials). Incubate 5 min in the dark.
- Seal the membrane in a plastic film. Put the membrane in an autoradiography cassette.
- Expose the membrane to an autoradiography film in the dedicated dark room.
NOTE: The exposition time depends on the signal strength, from few seconds to minutes. - Reveal the exposed film in an automatic developing device.
10. Preparation of the samples for RNA sequencing
NOTE: This step only concerns RNAs extracted from E and CE fractions.
- Add to each sample 10 µL of 10x DNase buffer and DNase I (1 U/µg of treated RNAs). Add water for a final volume of 100 µL.
- Incubate 1 h at 37 °C.
- Extract and purify RNAs as previously described (steps 8.2 to 8.11).
- Resuspend the RNA pellet in 20 µL of ultrapure water.
NOTE: The presence of remaining DNA can be checked using PCR and specific primers (e.g., 16S gene). - Assess RNA quantity and quality using a microfluidics-based electrophoresis analysis system (see Table of Materials).
NOTE: 1 µg is generally obtained in the elution fraction (E) after DNase treatment. - Remove ribosomal RNAs with a bacterial rRNA depletion kit.
NOTE: Large and abundant RNAs (i.e., rRNAs) tend to non-specifically interact with the affinity column. 500 ng of extracted RNA are required to perform this step. - Again assess RNA quantity and quality using a microfluidics-based electrophoresis analysis system.
- Prepare cDNA libraries with 10-20 ng of ribodepleted RNA using a cDNA library preparation kit and following manufacturer’s instructions.
- Sequence the obtained libraries using a sequencing instrument (e.g., single-end, 150 bp; see Table of Materials).
NOTE: 5-10 million reads per sample are generally enough.
11. RNAseq data analysis (Figure 2)
- Download the FastQ sequencing files from the sequencing platform.
- Access to the Galaxy instance of Roscoff biological station (https://galaxy.sb-roscoff.fr/) and log in.
NOTE: Every mentioned algorithm can be easily found using the search bar. A user guide is provided for each tool.
CAUTION: The version of required tools may differ from the public Galaxy server38. - Click on Get Data icon and then Upload File from your computer. Upload FastQ sequencing file of each MS2 control and MS2-sRNA samples. Upload also FASTA reference genome file and GFF annotation file.
- Run FastQC Read Quality reports (Galaxy Version 0.69).
NOTE: This tool provides a quality assessment of raw sequences (e.g., quality score, presence of adapter sequences). - Run Trimmomatic flexible read trimming tool (Galaxy Version 0.36.6) to notably remove adapter sequences and poor-quality reads. Indicate adapter sequences used for library preparation (e.g., TruSeq 3, single-ended). Add the following Trimmomatic operations: SLIDINGWINDOW (Number of bases=4; Average quality=20) and MINLEN (Min length of reads=20).
- Run again FastQC Read Quality reports (Galaxy Version 0.69).
- Run Bowtie2 – map reads against reference genome (Galaxy Version 2.3.2.2). Use the Genome Reference FASTA file from the history to map reads with default settings (very sensitive local).
NOTE: BAM file generated by Bowtie2 tool can be visualized using the Integrative Genomics Viewer (IGV). Associated BAI file will also be required. - Optionally, Run Flagstat which compiles stats for BAM dataset (Galaxy Version 2.0).
- Run htseq-count – Count (Galaxy Version 0.6.1) which aligns reads overlapping features in the GFF annotation file. Use the Intersection (non-empty) mode.
- Archive all raw counts files from htseq-count analysis into a single Zip file.
- Run SARTools DESeq2 to compare data (Galaxy Version 1.6.3.0). Provide the Zip file containing raw counts files and the design file, a tab delimited file describing the experiment. Carefully follow provided instructions to generate the design file.
The representative results originate from the study of RsaC targetome in S. aureus29. RsaC is an unconventional 1,116 nt-long sRNA. Its 5’ end contains several repeated regions while its 3’ end (544 nt) is structurally independent and contains all predicted interaction sites with its mRNA targets. The expression of this sRNA is induced when manganese (Mn) is scarce, which is often encountered in the context of host immune response. Using MAPS technology, we identified several mRNAs interacting directly with RsaC, revealing its crucial role in oxidative stress (sodA, ldh1 and sarA) and metal-related (znuBC-zur and sufCDSUB) responses.
Validation of the MS2-sRNA construct and experimental conditions
Before performing MAPS experiments, it is important to determine the optimum conditions of expression of the studied sRNA. If a non-native promoter is used, it will definitively help produce the MS2-sRNA construct when its targets are present. In addition, the MS2-sRNA construct should be carefully validated with regard to size, stability, expression and function. The MS2 aptamer was fused to the 5’ end of either the full-length RsaC (MS2-RsaC1116) or the shorter form (MS2-RsaC544) corresponding to the 3’ part of RsaC. Both constructs were expressed in vivo under the control of the quorum sensing dependent P3 promoter in S. aureus HG001 ΔrsaC. The deletion of rsaC gene avoids a competition between the endogenous RsaC and MS2-RsaC. The wild-type strain containing the same vector with the MS2 tag alone was used as control. This control allows subtracting unspecific interactions occurring with the MS2 tag.
To confirm the constructs and visualize their pattern of expression, bacterial cells were harvested after 2 h, 4 h and 6 h of growth in BHI medium at 37 °C. After RNA extraction, Northern blot analysis was performed using RsaC-specific DIG probe (Figure 3A). The level of endogenous RsaC (lanes 1-3) significantly increased after 6 h of growth, justifying the selection of this time point for MAPS experiments. Importantly, the levels of MS2-RsaC544 (lanes 7-9) and MS2-RsaC1,116 (lanes 10-12) were comparable to endogenous RsaC at 6 h. Hence, they should mimic the endogenous expression pattern of RsaC. A larger but minor form of RsaC was distinguishable and might be due to an inefficient end of transcription. This phenomenon is frequently observed when a MS2-sRNA is expressed under the control of a strong promoter from a plasmid21. No shorter forms resulting from aberrant transcription termination or degradation were observed.
The addition of the MS2 aptamer at the 5’ of sRNAs could also disrupt their proper folding and affect their functions. This step is critical for highly structured sRNAs as RsaC. Hence MS2-sRNA activity should be tested and compared to endogenous sRNA when possible. A previously known target or an observable phenotype can help to monitor it. For example, the impact of RsaC on intracellular ROS accumulation was used to validate MS2-RsaC544 and MS2-RsaC1,116 constructs29.
Analysis of collected fractions during affinity purification
RNAs were extracted from CE, FT and E fractions in WT strain expressing MS2 tag alone and ΔrsaC strain expressing MS2-RsaC544 construct. We showed using Northern blot analysis that the 1,116 nt-long endogenous RsaC was enriched in the elution fraction but turned out to interact non-specifically with the affinity column (Figure 3B, lanes 2-3). We observed the same phenomenon with MS2-RsaC1,116 (data not shown). This is certainly due to its length and complex secondary structure. Therefore, only a less structured and shorter form (544 nt) of RsaC corresponding to its 3’ part was used to perform MAPS experiments. In Figure 3B, the MS2-RsaC544 was highly enriched in the elution fraction demonstrating that it was successfully retained by the MS2-MBP fusion protein (lane 6). A larger but minor form of MS2-RsaC544 was observed as in Figure 3A. Here, the stringency and number of washes can be adjusted to either reduced non-specific binding or, on the contrary, to limit loss of true interacting partners.
Validation of putative mRNA targets after MAPS analysis
Following bioinformatic analysis, putative mRNA targets are listed according to the Fold-change between MS2-sRNA and MS2 control, obtained using DeSeq2 (Figure 2). For instance, MS2-RsaC544 MAPS data29 suggested that sodA mRNA, coding for a superoxide dismutase in S. aureus, is a main target (best hit, higher Fold-change). A Northern blot analysis, performed with a sodA-specific DIG probe after MS2-affinity purification, shows that sodA was efficiently co-enriched with MS2-RsaC544 compared to the MS2 control (Figure 3C).
A global transcriptomic analysis is systematically performed on the CE fraction. The comparison of MAPS data and this transcriptomic analysis helps adjust the enrichment ratio and reveals a potential target hierarchy. Indeed, a poorly expressed mRNA, which is highly enriched after MS2-affinity purification has certainly a greater binding affinity than a highly enriched and highly expressed mRNA.
It is important to note that all candidates identified by MAPS must be individually validated using in vitro and/or in vivo experiments such as Electrophoresis Mobility Shift Assays (EMSA) or reporter gene assays (see Jagodnik et al. (2017)39 for more details).
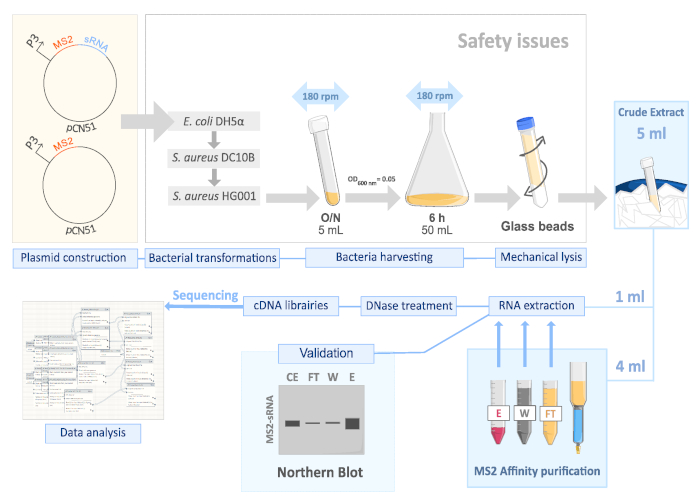
Figure 1. Schematic illustration of the MAPS protocol adapted to S. aureus. From plasmid construction to data analysis. Please click here to view a larger version of this figure.
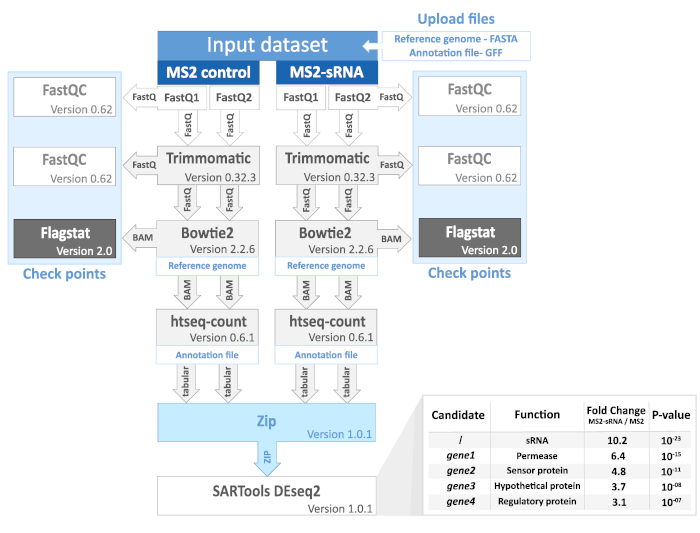
Figure 2. MAPS analysis workflow and processed data. Each step, check point and file format are represented (see also step 11). FastQ format is a text file consisting of the DNA sequences and corresponding quality scores. BAM format is a compressed file containing aligned sequences. Tabular format is a tab-delimited text file with counts for each gene. The results chart illustrates the kind of data obtained after bioinformatic analysis. Presented results are fictitious and do not originate from any study. For further details, basic tutorials are available on Galaxy Project website (https://galaxyproject.org/). Please click here to view a larger version of this figure.
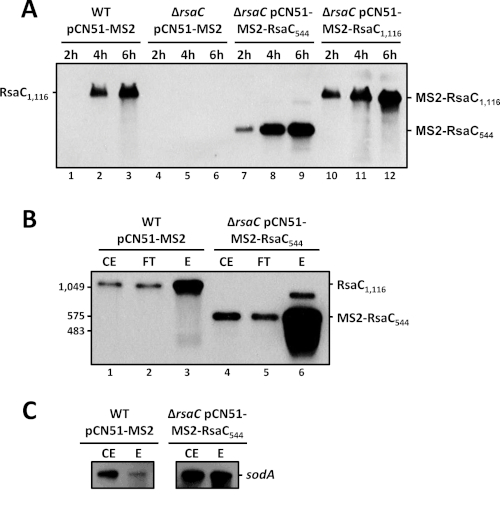
Figure 3: Constructs validation and MAPS controls. A. Northern blot analysis of endogenous RsaC sRNA and related MS2 constructs. WT strain carries the pCN51-P3-MS2 (control) and ΔrsaC mutant strain carry either the pCN51-P3-MS2, pCN51-P3-MS2-RsaC544 or pCN51-P3-MS2-RsaC1,116. Samples were taken after 2 h, 4 h and 6 h of growth in BHI at 37 °C. Northern blot assays were performed using a RsaC-specific DIG probe. B. Northern blot analysis of MS2-affinity purification fractions using a RsaC-specific DIG probe. The co-purification was performed using WT strain + pCN51-P3-MS2 (control) and ΔrsaC mutant strain + pCN51-P3-MS2-RsaC544. Cells were harvested after 6 h of growth in BHI at 37 °C. Crude extract (CE), flow-through (FT), elution (E). C. Northern blot analysis of MS2-affinity purification fractions (CE and E) using a sodA-specific DIG probe. See (B) for details. Please click here to view a larger version of this figure.
| RNA | Type | Reference |
| Escherichia coli | ||
| RyhB | sRNA | Lalaouna et al. (2015)21 |
| RybB | sRNA | Lalaouna et al. (2015)21 |
| 3'ETSleuZ | tRNA-derived fragment | Lalaouna and Massé (2015)26 |
| DsrA | sRNA | Lalaouna et al. (2015)22 |
| hns | mRNA (5'UTR) | Lalaouna et al. (2015)22 |
| CyaR | sRNA | Lalaouna et al. (2018)23 |
| RprA | sRNA | Lalaouna et al. (2018)23 |
| GcvB | sRNA | Lalaouna et al. (2019)24 |
| Salmonella Typhimurium | ||
| SraL | sRNA | Silva et al. (2019)25 |
| Staphylococcus aureus | ||
| RsaA | sRNA | Tomasini et al. (2017)27 |
| RsaC | sRNA | Lalaouna et al. (2019)29 |
| RsaI | sRNA | Bronesky et al. (2019)28 |
Table 1. MAPS technology revealed the targetome of several RNAs in various organisms.
