Quantification of tubulin in the P2, P3, and S3 fractions from mouse brain by the MT fractionation method
Tubulin in mouse tissue was separated into the P2, P3, and S3 fractions by the MT fractionation method and quantified by Western blotting (Figure 1A). The precipitate of MTs that remained in the P2 fraction by ultracentrifugation at 100,000 × g for 20 min accounted for 34.86% ± 1.68% of total tubulin in a mouse brain. The supernatant (S2) was further centrifuged at 500,000 × g for 60 min. A precipitate (P3 fraction) and a supernatant (S3 fraction) were obtained, which accounted for 56.13% ± 2.12% or 9.01% ± 0.68% of total tubulin in the mouse brains, respectively (Figure 1B).
The cerebral cortex used in this study contains neuronal and nonneuronal cells, such as glia. To selectively assess MT stability in the neurons of mouse brains, we quantified TUBB3, a tubulin subtype exclusively expressed in neurons in the central and peripheral nervous system, by Western blotting with Tuj1 (anti-TUBB3 antibody). The percentage of TUBB3 in the P2, P3, or S3 fraction was 32.65% ± 2.20%, 59.31% ± 2.61%, or 8.04% ± 0.74%, respectively. They were not significantly different from those of α-tubulin (Figure 1B). These results suggested that neurons and glial cells exhibit similar MT stability or that neurons contain much higher amounts of tubulin than that of glial cells in vivo.
Characteristics of tubulin recovered in each fraction
Here, the mouse tissue homogenate was separated into three fractions by two-step ultracentrifugation with different gravitational accelerations; therefore, the sedimentation coefficients among proteins or their complexes in each fraction differed. Although tubulin precipitated at 100,000 × g ultracentrifugation was considered conventional MT, how the newly obtained tubulin of the P3 fraction here differs from that of tubulin in the P2 and S3 fractions should be clarified.
To characterize S3 tubulin, the S2 (P3 + S3) or S3 fraction was subjected to ultrafiltration and size exclusion chromatography. The tubulin complexes in the S3 fraction could pass through a 300 kDa ultrafiltration spin column completely, while almost all tubulin in the S2 fraction was trapped on the filter (Figure 2A). Furthermore, the molecular weight of tubulin complexes in the S3 fraction was measured by size exclusion chromatography. S3 tubulin eluted at one peak corresponding to 100 kDa, which is similar to that of commercially available purified tubulin dimers (Figure 2B,C). In addition, the proportions of α- and β-tubulin recovered in each fraction by the MT fractionation method were equal (Figure 2D). Actually, it has been shown that α- and β-tubulin can slightly exist as monomers in living cells24. However, judging from the estimated kD value (nM order) reported, and the concentration of tubulin recovered in the S3 fraction (~11 µM), most (> 98%) tubulin is thought to exist as α/β-dimers. Therefore, tubulin in the S3 fraction is primarily a soluble α/β-tubulin dimer.
The tubulin polymers were separated into two fractions, P2 and P3, based on their post-translational modifications (PTMs). To differentiate between these fractions, Western blotting was performed using specific antibodies. The anti-acetylated α-tubulin antibody, which serves as a marker for stable MTs, demonstrated that the P2 fraction was significantly enriched with 97.40% ± 0.52% of acetylated α-tubulin (Figure 2E), while the total α-tubulin was recovered in the P3 and S3 fractions (Figure 1B). Conversely, the anti-tyrosinated α-tubulin antibody, indicative of labile MTs, revealed that 75.43% ± 2.69% of tyrosinated α-tubulin was present in the P3 fraction (Figure 2F). These findings confirm that the P2 fraction primarily contains tubulin within stable MTs, whereas the P3 fraction consists of tubulin within labile MTs.
Assessment of MT stability under freezing and nocodazole treatment
The effect of freezing and nocodazole treatment on the stability of mouse intracerebral MTs was analyzed to determine whether transient changes in the stability of MTs could be discerned by the MT fractionation method. MTs generally disassemble at low temperatures, but some MTs remain stable in the cold. After weighing the brains, they were frozen in liquid nitrogen and placed at -80 °C for 30 min. The transient frozen brain and raw brain as control were homogenized and fractionated into the P2, P3, and S3 fractions. Then, the proportion of tubulin contained in the three fractions was quantified by Western blotting. Once the brain was frozen before homogenization, α-tubulin in the P2 fraction decreased, and that in the P3 fraction increased in comparison to that in the raw brain (Figure 3A). Blots with 6-11B1 (acetylated α-tubulin) or 1A2 (tyrosinated α-tubulin) also revealed that brain freezing decreased the acetylation level (Figure 3B) and increased the tyrosination level (Figure 3C) of α-tubulin in the P2 fraction.
Nocodazole is a microtubule-targeting agent (MTA) that prevents MT polymerization by binding to β-tubulin and promotes MT depolymerization. Mouse brains were homogenized in Taxol-free MSB(+) with or without 10 µM nocodazole and placed at 4 °C for 20 min. The nontreated or nocodazole-treated homogenate was added to 10 µM Taxol, re-homogenized, and fractionated into the P2, P3, and S3 fractions. Then, the proportion of tubulin contained in the three fractions was quantified by Western blotting. Blots with DM1A (α-tubulin) showed that α-tubulin in the P2 fraction decreased and that in the P3 fraction tended to increase with nocodazole treatment (Figure 3D). These results indicate that P2 tubulin was destabilized in response to low temperature or nocodazole and that the P2 fraction contained robust MTs that resisted the experimental conditions. In contrast to freezing, nocodazole did not affect tubulin PTMs (Figure 3E,F). Based on the results and the above data, the depolymerization of MTs that cannot be detected by PTM analysis can be evaluated by this method.
Comparison of the ratio of stable MTs, labile MTs, and free tubulin in tissues
The stability of MTs varies among different tissues, depending on the proliferative capacity of the cells within those tissues. Notably, stable MTs are more abundant in the nervous system, which primarily consists of nonproliferative neurons, compared to other tissues4. To assess the ability of the developed MT fractionation method to discern differences in MT stability across various tissues, the livers and thymi of mice were fractionated, and the recovered tubulin in each fraction was quantified. The results revealed that, in comparison to other tissues, the brain exhibited a significantly higher level of P2 tubulins, while P3 tubulins were notably enriched in tissues containing proliferative cells (Figure 4). Additionally, Western blotting using 6-11B1 and 1A2 antibodies confirmed the presence of higher MT stability in the nervous system. The distinct distribution patterns of tubulin PTMs clearly indicated that the P2 tubulin specifically found in the nervous system originated from stable MTs (Figure 4). These findings further support the notion that the P2 and P3 fractions correspond to stable and labile MTs, respectively.
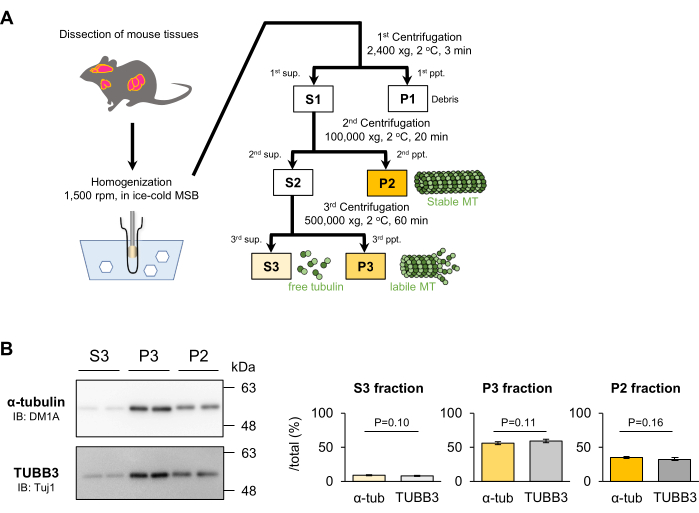
Figure 1: Quantification of tubulin in mouse tissue using the MT fractionation method. (A) Summary of the MT fractionation method for tissues. Stable MTs (P2 fraction), labile MTs (P3 fraction), and free tubulin (S3 fraction) in tissues can be separated by 2-step ultracentrifugation under conditions that suppress MT polymerization and depolymerization during preparation. (B) MTs in the mouse cortex were precipitated by conventional 100,000 × g ultracentrifugation followed by 500,000 × g ultracentrifugation. Then, tubulins separated into the P2, P3, and S3 fractions were quantified by Western blotting with DM1A (α-tubulin) and anti-Tuj1 (TUBB3). The proportion of proteins in each fraction (P2, P3, or S3) to the total fraction (P2 + P3 + S3) was calculated as described in the Protocol section (means ± SDs, n = 4). This figure has been modified from Hagita et al.23. Please click here to view a larger version of this figure.
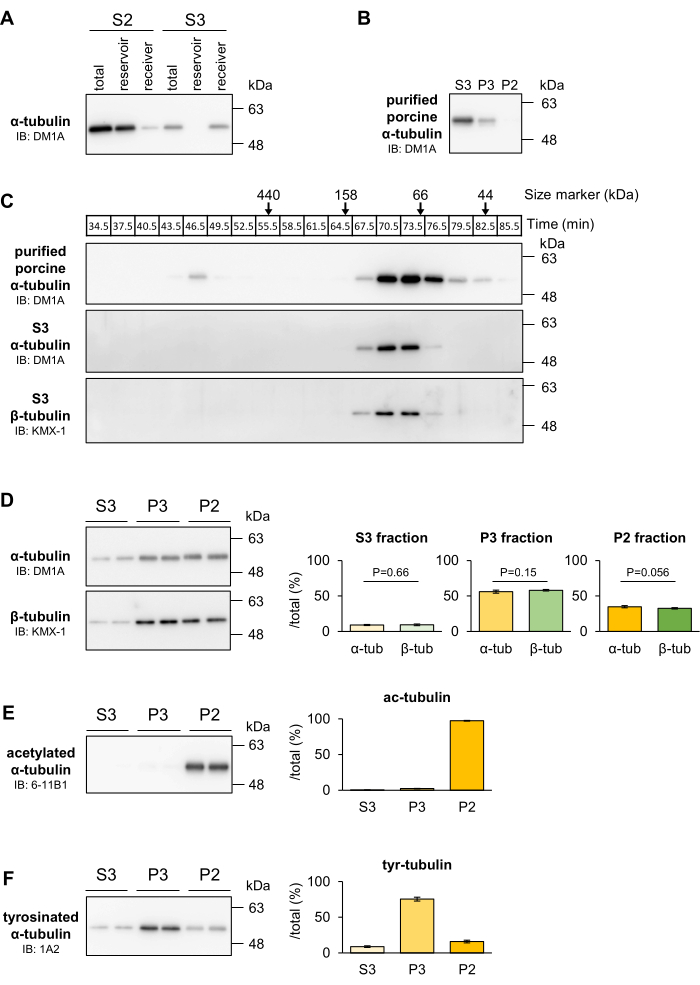
Figure 2: The MT fractionation method enables the separation of stable MTs, labile MTs, and free tubulin in the mouse cerebral cortex. (A) The S2 or S3 fractions (input) were analyzed by the filter trap assay using a 300 kDa ultrafiltration spin column. Tubulins obtained by filtration (receiver) and trapping (reservoir) were quantified by Western blotting with DM1A (α-tubulin). (B) Purified porcine tubulins were subjected to the MT fractionation method and were found to be mostly collected in the S3 fraction. (C) Molecular size of tubulin in the S3 fraction. The S3 fraction was separated by size exclusion chromatography using a gel filtration chromatography column. Proteins in each fraction were quantified by Western blotting with DM1A (α-tubulin) and KMX-1 (β-tubulin). The theoretical molecular weight is shown at the top of the panels. (D) The proportions of α-tubulin and β-tubulin in each fraction were equal. Tubulins separated into the P2, P3, and S3 fractions were quantified by Western blotting with KMX-1 (β-tubulin). Quantification of the proportion of α-tubulin and β-tubulin in each fraction relative to the sum of the total fraction (means ± SDs, n = 4). Statistical analyses were performed by Student's t-test. (E,F) The modifications of α-tubulin in the P2, P3, and S3 fractions were verified by Western blotting with 6-11B1 (acetylated α-tubulin: E) and 1A2 (tyrosinated α-tubulin: F). Quantification of the proportion of tyrosinated or acetylated α-tubulin in each fraction relative to the total fraction (means ± SDs, n = 4). (A–C) have been modified from Hagita et al.23. Please click here to view a larger version of this figure.
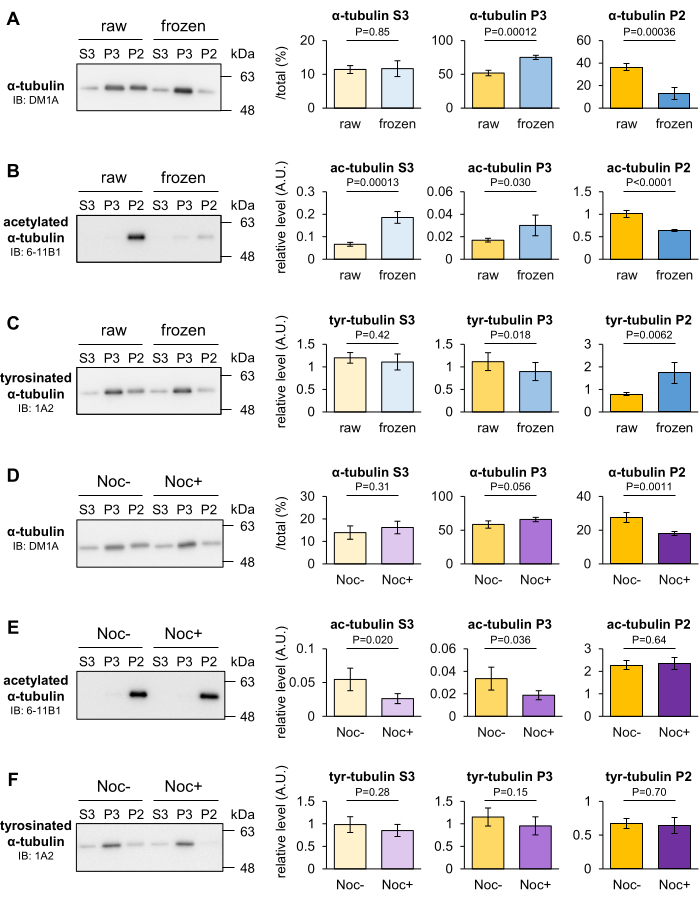
Figure 3: Assessment of MT depolymerization induced by freezing or MTA. (A–C) The stability of MT after 30 min of freezing at -80 °C was evaluated. The proportions of tubulin contained in the three fractions of the raw and frozen brain were quantified by Western blotting with DM1A (α-tubulin: A), 1A2 (tyrosinated α-tubulin: B), and 6-11B1 (acetylated α-tubulin: C). (D–F) The effect of nocodazole on MT stability was evaluated. The proportions of tubulin contained in the three fractions of nocodazole untreated or treated brains were quantified by Western blotting with DM1A (α-tubulin: D), 1A2 (tyrosinated α-tubulin: E), and 6-11B1 (acetylated α-tubulin: F). Representative relative levels of tyrosination (B,E) or acetylation (C,F) were normalized to the amount of total α-tubulin (A,D) (means ± SDs, n = 4). Statistical analyses were performed by Student's t-test. Please click here to view a larger version of this figure.
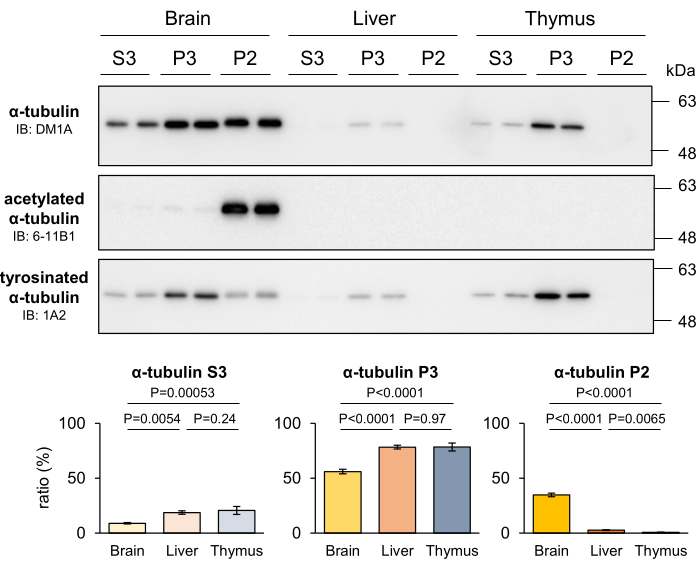
Figure 4: Proportions of stable MTs, labile MTs, and free tubulin in the brain, liver, and thymus in mouse. The brains, livers, and thymi of mice were dissected and subjected to the MT fractionation method. Proteins separated into each fraction were quantified by Western blotting with DM1A (α-tubulin), 6-11B1 (acetylated α-tubulin), and 1A2 (tyrosinated α-tubulin). The proportion of proteins in each fraction (P2, P3, or S3) to the total fraction (P2 + P3 + S3) was calculated as described in the Protocol section (means ± SDs, n = 4). Statistical analyses were performed by one-way ANOVA followed by Tukey's post hoc test. Please click here to view a larger version of this figure.
