We have presented a comprehensive SPA pipeline to obtain a high-resolution structure using three different processing platforms: cryoSPARC v3, RELION-3, and Scipion 3. Figure 1 and Figure 4 summarize the general processing workflow, and Table 1 details refinement protocols. These protocols were used during refinements of a 2.3 Å structure of AAV, achieving near Nyquist resolution.
Movies were first imported to cryoSPARC v3 and subsequently motion- and CTF-corrected to generate averaged micrographs. When selecting micrographs for further processing, it is important to choose those with a good CTF-fit and low astigmatism (Figure 2), as including poor-quality micrographs can hinder later processing stages, resulting in a lower resolution reconstruction. 27,364 particles were then picked and extracted from the selected micrographs. Because the diameter of the AAV is approximately 220 Å and pixel size is 1.045 Å, a box size of 300 px was used. Next, iterative 2D classification was used to remove artifacts and particles not converging to stable classes. Examples of selected and excluded 2D class averages are presented in Figure 3. It is also important to note that class averages reflecting different conformations of the specimen should be refined separately to yield multiple 3D reconstructions. In such a case, multiple ab initio starting volumes should be calculated. Here, 26,741 particles were selected and used for ab initio modeling and homogeneous refinement of a single 2.9 Å structure.
After transferring coordinates of particles picked in cryoSPARC v3 to RELION-3, we carried out four additional rounds of 2D classification until the data set converged to stable 2D classes. The above-described 2D classification removed an additional 3,154 particles from the data set. Using the structure generated in cryoSPARC v3 as an initial model, 3D refinement in RELION-3 produced a structure with a nearly equivalent resolution of 2.95 Å. Subsequent structural refinements, which included per-particle motion correction and CTF refinements, increased the resolution to 2.61 Å. A complete list of refinements we performed is presented in Table 1. The volume calculated in RELION-3 was then further refined in Scipion 3 using multiple rounds of high-resolution refinement (Xmipp3 – highres). During subsequent rounds of refinement, an additional 3,186 particles were removed from the data set, resulting in a final set of 20,401 particles, which produced a 2.3 Å reconstruction of AAV (Figure 5 and Figure 6). Thus, given the pixel size of 1.045 Å, our refinements have nearly reached the Nyquist limit. FSC curves representing structures calculated using each program are shown in Figure 6. These FSC curves indicate the resolution increase throughout the workflow. Because resolution may vary from point to point in the map, it is often more appropriate to present the distribution of local resolution estimates in the map rather than reporting a single resolution estimate according to a single criterion (e.g., 0.143 criterion) from the FSC curve. Thus, we performed such analysis using Xmipp – MonoRes in Scipion 3. Figure 7 shows a comparison of local resolution estimates for maps obtained with cryoSPARC v3 and Scipion 3. Resolution estimates at four different slices through the structures (Figure 7A,B) and resolution histograms (Figure 7C) clearly demonstrate the incremental improvement in local resolution between the maps throughout the workflow. The FSC curve calculated using the program Xmipp3 – highres in Scipion 3 indicates the Nyquist limit has been reached (Figure 6), suggesting the resolution estimate is very likely limited by undersampling29. However, MonoRes analysis presented in Figure 7C, along with a careful analysis of the EM map and map fitting with atomic coordinates of AAV (Figure 5) suggest that a more adequate resolution estimate for the map is 2.3 Å. A similar strategy reconciling the FSC and MonoRes resolution estimates have been presented earlier24,25. Because resolution estimates can be influenced by the mask used during refinement steps, it is important to ensure the mask does not exclude any part of the density. The mask used in this study overlapped with the 3D reconstructions is presented in Figure 7D. The gradual increase in resolution in the presented workflow highlights the advantage of utilizing algorithms from multiple SPA software packages to achieve a high-quality and high-resolution 3D reconstruction.
In-situ model building or fitting the map with a pre-existing atomic model can serve as the quality check for the calculated structure. We have visualized the final map in UCSF Chimera and fitted the map with a previously published atomic model (PDB ID: 7kfr)30. Figure 5 shows regions of the cryo-EM map fitted with atomic coordinates of AAV. Well-defined EM densities allow for fitting side-chains of individual amino acids, water molecules, and magnesium ions and confirm the agreement of the cryo-EM map with the atomic model.

Figure 1: Complete SPA workflow across cryoSPARC v3, RELION-3, Scipion 3, and Phenix 1.18. Steps completed in cryoSPARC v3, RELION-3, Scipion 3, and Phenix 1.18 are denoted with purple, orange, green, and grey boxes, respectively. The time required for completion of each step using the processing server equipped with 8 GPUs, 40 CPUs and 750 GB of RAM is specified in each individual box. Please click here to view a larger version of this figure.

Figure 2: Selection of micrographs for downstream processing in cryoSPARC v3. (A) Micrographs with well-matched estimated and experimental Thon rings were used for further processing, while those with high astigmatism and poor fit (B) were discarded. Micrographs with CTF-fit above 5 Å, astigmatism over 400 Å, and relative ice thickness below 2 were removed from further processing, i.e., 70/395 micrographs. Please click here to view a larger version of this figure.
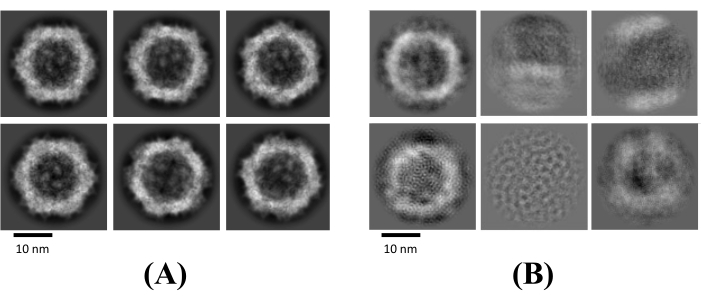
Figure 3: Selecting 2D classes. 2D class averages containing well-defined classes are selected (A), and those with low-resolution, noise, and partial particles are rejected (B). Please click here to view a larger version of this figure.
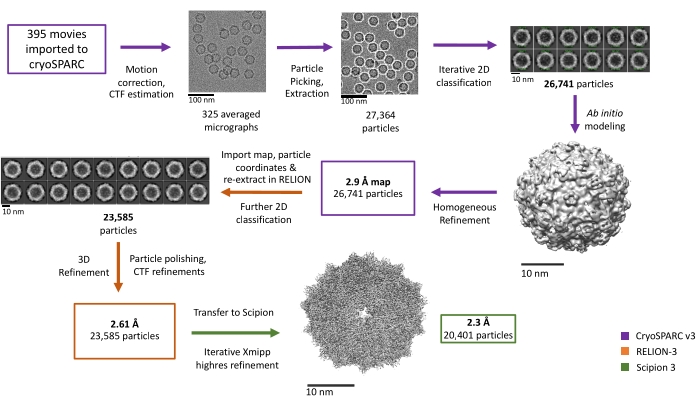
Figure 4: Workflow and representative results for AAV processing across cryoSPARC v3, RELION-3, and Scipion 3. Steps completed in cryoSPARC v3, RELION-3, and Scipion 3 are denoted with purple, orange, and green arrows, respectively. Please click here to view a larger version of this figure.

Figure 5: High-resolution structure of AAV shows well-defined EM densities representing different secondary structure elements and individual amino acid side chains. (A) A final map of AAV. (B) A part of the map representing beta sheets fitted with atomic coordinates of AAV (PDB ID: 7kfr)30. (C) Map densities representing individual amino acids. From left to right: arginine, phenylalanine, and tryptophan. (D) High-resolution features of the map include water molecules presented in red and magnesium ions presented as green spheres. Mg2+ ion displayed in the figure is coordinated by histidine (left) and arginine residues. Please click here to view a larger version of this figure.
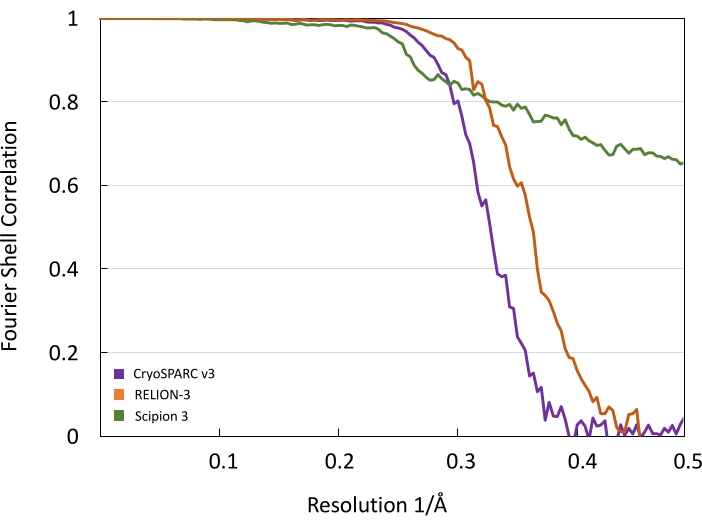
Figure 6: FSC curves from cryoSPARC v3, RELION-3, and Scipion 3 show increasing resolution across the workflow. While the FSC curve calculated using the program Xmipp3 – highres in Scipion 3 indicates the Nyquist limit has been reached, suggesting the resolution estimate is limited by undersampling29, a more adequate analysis of the map resolution is presented in Figure 7 and discussed in the Representative Results section24,25. Please click here to view a larger version of this figure.
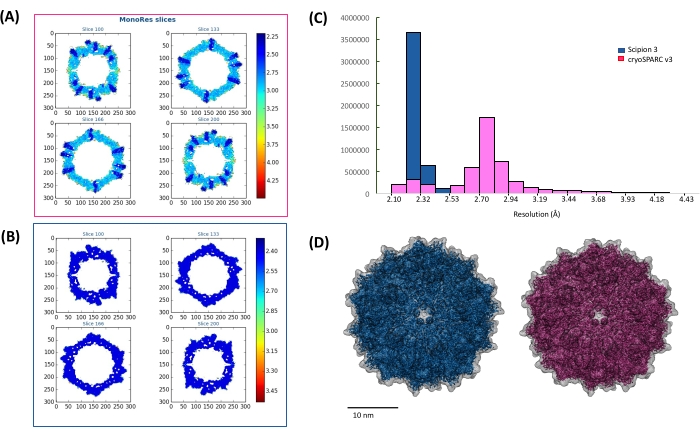
Figure 7: Validating the final reconstruction in Scipion 3 using Xmipp – MonoRes. Resolution of the map is better described by presenting local resolution distributions rather than a single resolution estimate according to a single criterion from the FSC curve. (A–B) Panels A and B show different slices from maps generated in cryoSPARC v3 and Scipion 3, respectively. (C) Histograms demonstrating a systematic increase in local resolution for maps calculated in cryoSPARC v3 (pink bars) and Scipion 3 (blue bars). (D) The mask (gray) used for local resolution calculations contain all parts of the AAV densities refined in both programs. Please click here to view a larger version of this figure.
| Program | Refinement type | Script | |||
| cryoSPARC v3 | Homogeneous Refinement | Homogeneous Refinement | |||
| Non-uniform Refinement | Nonuniform Refinement | ||||
| Heterogeneous Refinement | Heterogeneous Refinement | ||||
| Per-particle motion correction | Local Motion Correction | ||||
| RELION-3 | 3D refinement | Refine3D | |||
| Postprocessing – B-factor Sharpening, MTF Correction | Refine3D | ||||
| Particle Polishing | Bayesian Polishing | ||||
| CTF refinement – beam tilt | CtfRefine | ||||
| CTF refinement – anisotropic magnification | CtfRefine | ||||
| CTF refinement – per-particle defocus, per-particle/micrograph astigmatism | CtfRefine | ||||
| Ewald sphere curvature correction | Relion_reconstruct | ||||
| Scipion 3 | High resolution refinement | Xmipp3 – highres | |||
| Phenix 1.18 | Density Modification and Sharpening | ResolveCryoEM | |||
Table 1: Refinements implemented throughout the workflow. Whether certain refinements are applicable to a specific project depends on data quality and acquisition parameters. For example, the Ewald sphere curvature correction can be applied for maps that already have high resolution.
