Primary liver cells, enriched for hepatocytes, were isolated from the mouse liver by two-step collagenase perfusion, using a peristaltic pump to circulate warm buffers through the liver taking advantage of the organ's vasculature to deliver dissociation enzymes to all cells (Figure 1A). For this, the inferior vena cava was cannulated, and the portal vein was snipped to allow the flow-through of buffers (Figure 1B). First, an HBSS-based buffer was flushed through the liver to clear the blood. If the cannulation is successful and there are no blood clots, the liver blanches and becomes yellow within a few seconds. Secondly, a Digestion buffer containing the Liberase enzyme blend was circulated through the liver to dissociate the tissue into a single-cell suspension. The cells were manually counted and seeded into 96-well ultra-low-adherence (ULA) plates, which enable the self-assembly into spheroids within a few days. On day 5, the spheroids are formed, and the thin capsule bordering the spheroids indicates successful aggregation (Figure 1C). We wait until day 10 to transplant, at which point the spheroids are compact and have developed strong cell-cell connections. The number of seeded cells per well determined the size of the liver spheroid, with 1000, 1200, and 1500 cells/well yielding spheroids of 238 µm ± 10 µm, 248 µm ± 17 µm, and 298 µm ± 19 µm (mean ± SD) diameters, respectively (Figure 1C,D). For transplantation, we select spheroids of approximately 250 µm diameter for the following reasons: (1) the spheroids size should not be too large to avoid hypoxia and necrotic core, but should contain enough cells to support cell-cell communications and to allow graft remodeling in the eye, (2) the weight of spheroids of this size allows them to gravitate towards the iris and improve their engraftment, (3) this size is appropriate in relation to transplanting 5-10 spheroids per mouse eye.
The transplantation surgery requires a manual-threaded syringe connected to a glass cannula (Figure 2A). The glass cannula consists of a borosilicate glass capillary modified in-house to have a fine blunt tip using a micropipette puller and beveler. A simpler alternative cannula can be created using a commercially available plastic catheter connected to the syringe tubing and stabilized in a pipette tip (Figure 2B). The surgery consists of the inoculation of liver spheroids into the ACE through an incision in the cornea (Figure 2C). The spheroids were positioned on the borders of the pupil to make them better accessible for imaging and avoid them moving into the ocular angle. Albino mice were used for transplantation, as their non-pigmented iris allow the in vivo imaging of the engrafted liver spheroids. Recipient mice were transplanted into both eyes with 7-10 spheroids/eye, and stereoscopic images were taken at 3 days post-transplantation (post-Tx) as well as at 1 week and 1 month post-Tx to document the cornea healing and spheroid engraftment success (Figure 2D). Of note, the change in appearance of the liver spheroids in the ACE between when freshly transplanted and fully engrafted is due to the settlement of the graft onto the iris, as well as to the growth of a monolayer of iris cells over the spheroid. The engraftment success rate of liver spheroids in the ACE is 70% (n = 9 eyes in both male and female mice) (Figure 2E). The first days post-Tx are the most critical for survival and engraftment, likely due to the recipient animal rubbing its eyes and dislodging the spheroids before the cornea has healed. The size of the liver spheroids does not differ significantly post-Tx and changes to shape are attributed to graft remodeling and engraftment (Figure 2F). At 1 month post-Tx, all engrafted spheroids present on the iris were vascularized and innervated, as shown by immunofluorescence staining (Figure 2G).
Noninvasive in vivo imaging is performed on anesthetized recipient mice using an upright confocal microscope and long-distance dipping objective (Figure 3A, Table 2). The fluorescence imaging in the ACE can be achieved through different approaches, as depicted in Figure 3B. The injection of fluorescent probes into the circulation of the recipient mouse allows the visualization of different cell types and structures within the spheroids. We used lectin to mark blood vessels (Figure 3C), CMFDA to observe the bile canaliculi network (Figure 3D) and pHrodo-LDL, which confirmed active LDL-uptake into spheroid cells (Figure 3E). Liver spheroids generated from reporter mouse models can also be used. Albumin-Cre:tdTomato spheroids allowed labeling and tracking of hepatocytes (Figure 3F), and spheroids expressing the Fluorescent Ubiquitin Cell Cycle Indicator (FUCCI) biosensor were used to visualize cell cycle dynamics at single-cell resolution (Figure 3G). Finally, liver spheroids can be genetically modified in vitro prior to transplantation, and, in the case of adeno-associated virus (AAV)-GFP transduction, the expression was observed in vivo for over 6 months (Figure 3H).
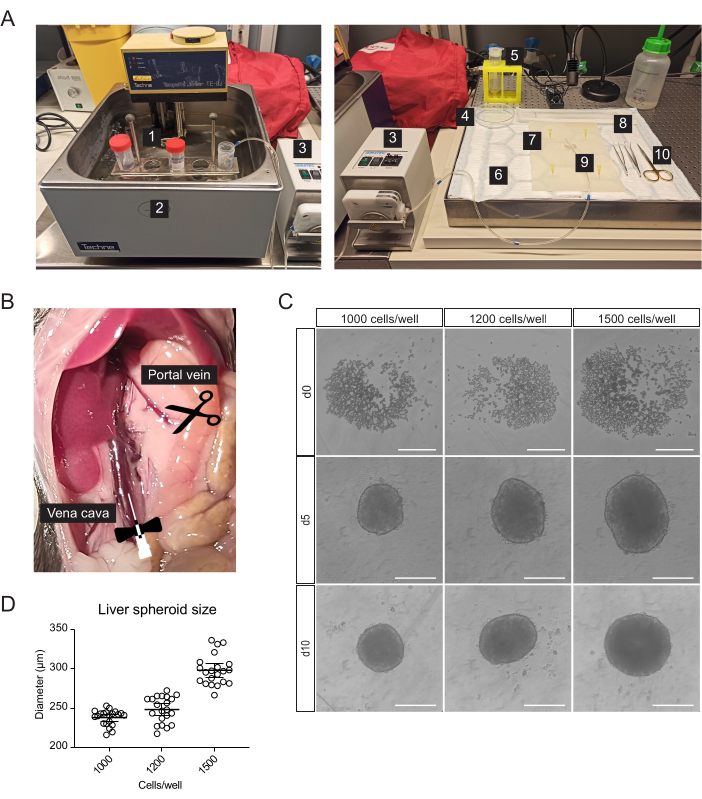
Figure 1: Isolation of primary mouse hepatocytes and generation of liver spheroids. (A) Material and equipment used for isolation of primary mouse hepatocytes: 1. Isolation buffers; 2. Water bath; 3. Peristaltic pump; 4. Petri dish; 5. Cell strainer; 6. Absorbent pad; 7. Dissection mat; 8. Cell Lifter; 9. Butterfly needle 27 G; 10. Dissection tools. (B) Abdominal cavity during surgery: the vena cava is cannulated and perfused, and the portal vein is snipped to allow flow-through of the buffers. (C) Brightfield images of the formation of hepatic spheroids in vitro at 0 (d0), 5 (d5), and 10 (d10) days post-seeding, scale bars = 200 µm. (D) Liver spheroid size at different cell-seeding concentrations, n = 21 spheroids. Please click here to view a larger version of this figure.
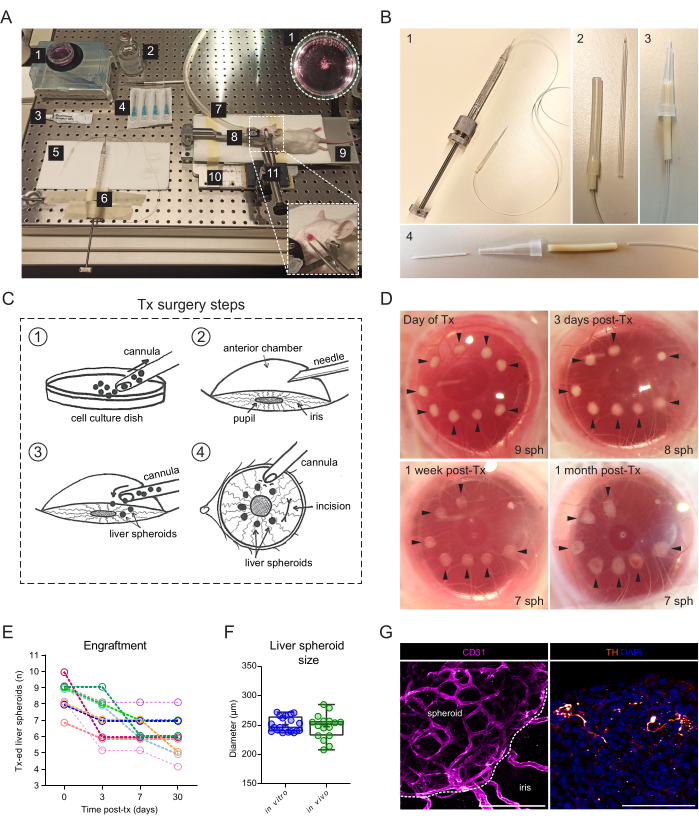
Figure 2: Transplantation and engraftment of liver spheroids into the ACE of mice. (A) Materials and equipment used for transplantation (Tx) of liver spheroids into the ACE: 1. Liver spheroids in culture dish; 2. Sterile saline; 3. Eye ointment; 4. Needles 23 G; 5. Cannula; 6. Hamilton syringe; 7. Anesthesia gas tube; 8. Head-holder and gas mask; 9. Heating pad; 10. Custom-made metal base plate; 11. Forceps and solid universal joint. (B) Cannula and Hamilton syringe setup: 1. Glass cannula connected to the Hamilton syringe via Portex tubing and a 27G needle; 2. Glass cannula is connected to the Portex tubing via additional segments of Silicone tubing and PharMed tubing; 3. Alternative assembled plastic cannula; 4. Parts forming the plastic cannula: 24G BD Insyte plastic catheter connected via PharMed tubing and sheathed in a cut-off 10 µl pipette tip for stability and grip. (C) Illustration of Tx surgery steps: 1. The spheroids are collected into the cannula; 2. The cornea is punctured with a needle; 3. The cannula is inserted into the incision, and the spheroids are released into the ACE; 4. From the outside of the eye, the spheroids are positioned close to the pupil and away from the incision. (D) Stereoscopic images of liver spheroids (sph) in the mouse eye on the day of surgery and at 3-, 7-, and 30-days post-Tx. Arrows indicate viable spheroids. (E) Liver spheroid (size of 1200 cells/well) engraftment rate post-Tx, n= 9 eyes in 6 recipient mice. (F) Size of liver spheroids in culture, prior to transplantation (in vitro, n= 20 spheroids from single preparation) and at 1-month post-Tx in the ACE (in vivo, n= 16 spheroids in 3 recipient mice), calculated by averaging vertical and horizontal diameters. (G) Immunofluorescence staining of engrafted liver spheroids at 2 months post-Tx, showing vascularization (CD31, pink, dashed line delineates the spheroid mass) and sympathetic innervation (tyrosine hydroxylase (TH), orange), scale bar = 100 µm. Data for panel F has been adapted with permission from Lazzeri-Barcelo et al.10. Please click here to view a larger version of this figure.
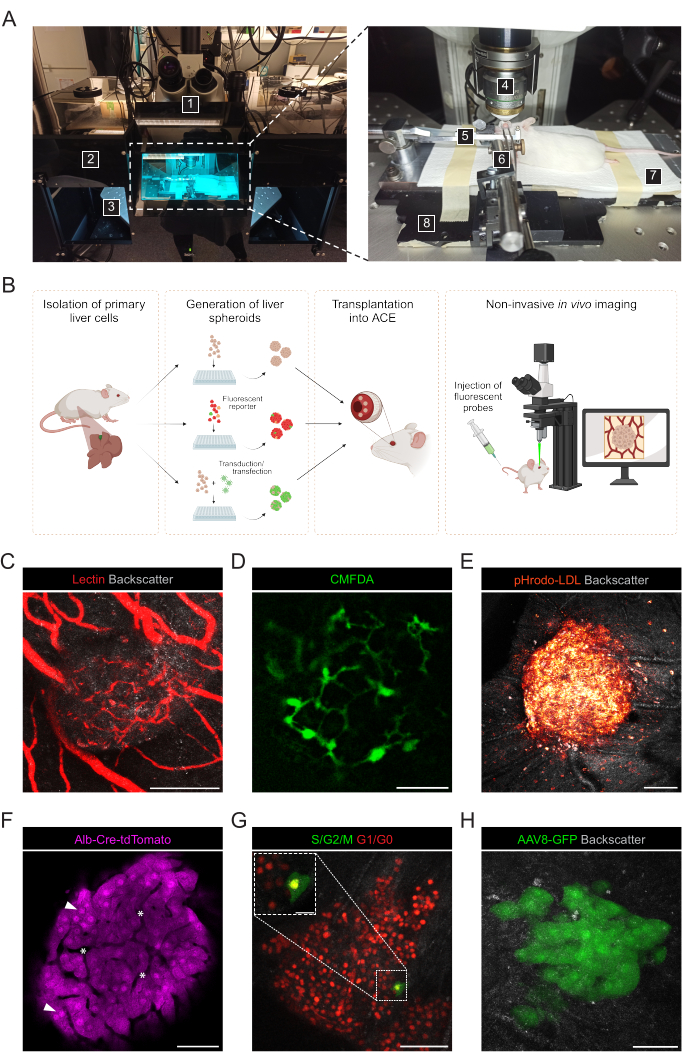
Figure 3: Noninvasive intraocular in vivo imaging of engrafted liver spheroids. (A) Material and equipment used for in vivo ACE imaging: 1. Upright laser scanning confocal microscope; 2. Dark box; 3. Motorized XYZ stage; 4. Dipping-objective; 5. Head-holder and gas mask; 6. Forceps and solid universal joint; 7. Heating pad; 8. Custom-made metal base plate. (B) Diagram depicting different approaches used for in vivo imaging of fluorescent readouts in liver spheroids engrafted in the eye. (C-H) Representative images of ACE-liver spheroids during in vivo imaging by confocal microscopy. The backscatter signal is used to observe the spheroid volume and structure; (C) Blood vessels labeled by i.v. injection of fluorescent lectin, scale bar = 100 µm; (D) Bile canaliculi network labeled by injection of fluorescent CMFDA, scale bar = 50 µm; (E) LDL uptake by injection of fluorescent pHrodo-LDL probe, scale bar= 100 µm; (F) Td-Tomato-expressing hepatocytes, arrowheads indicate nuclei and asterisks indicate intra-spheroid vasculature, scale bar = 50 µm; (G) Monitoring of cell cycle dynamics in FUCCI-expressing liver spheroids, scale bars = 50 µm (main image) and 20 µm (blow-up). (H) liver spheroids transduced in vitro with AAV8-GFP prior to Tx and imaged in the eye at 6 months post-Tx, scale bar = 50 µm. The image in panel G has been adapted with permission from Lazzeri-Barcelo et al.10. Please click here to view a larger version of this figure.
Table 1: Solutions used for the isolation of primary mouse hepatocytes. Composition of solutions and buffers needed for mouse hepatocyte isolation. The Digestion buffer and Gradient solution components should be mixed fresh on the day of isolation. Please click here to download this Table.
Table 2. Confocal Leica SP5 microscope settings used for intraocular in vivo imaging of liver spheroids. The table has been adapted with permission from Lazzeri et al.10. Please click here to download this Table.
