General Comments on LCM of Drosophila Peripheral Neurons
Allow from 6 hours, up to a week or longer for LCM depending upon the tissue type and the number of cells required.
All the procedures are carried out in strictly RNAse-free conditions following standard procedures. Larvae expressing either 21-7-GAL4,UAS-mCD8::GFP or ppk-GAL4,UAS-mCD8::GFP transgenic reporter lines were used for these experiments.
1. Preparing the larvae
- Collect 30-40 age-matched third instar larvae and wash them in ddH2O followed by brief rinses in RNAse Away (Sigma-Aldrich), and a final wash in ddH2O to remove RNAse Away. Wick off the excess ddH2O completely with a clean Kimwipe prior to embedding the larvae in OCT.
- Take a clean tissue embedding mold and layer it with a thin (1.5-2mm) layer of OCT, just enough to cover a single layer of larvae.
- Prior to embedding the larvae, if needed, cool the OCT to 0°C in the tissue embedding molds. Do this by placing the mold containing OCT on a block of ice. This will help reduce larval movement during the embedding process.
- Place the clean larvae on the pre-cooled OCT and arrange them in parallel to one another. Once the larvae have been arranged, slowly fill the mold with OCT without disturbing the larval arrangement. Snap freeze the mold by placing it on a dry ice block. [Critical Step: Snap-freeze the larvae instantly to reduce movement.] This method will allow the larvae to be in a single plane, maximizing the number of cells available per section for capture.
If it is necessary to preserve the tissue morphology for identifying the cells of interest, or if the expected number of cells per section is found to be satisfactory then proceed directly to Step 11.
Alternatively if the cells are labeled specifically and can be identified with an easily identifiable marker such as GFP or RFP, but the number of cells per section is found to be low, then Steps 5-10 may be helpful for increasing the number of cells available for capture per section. These are optional steps and should be considered only if required, as a method for increasing the number of da neurons available for LCM per section when the other method fails to provide favorable results. [Caution: This method may disrupt the overall tissue morphology and make tissue identification based on specific spatial location very difficult.]
- Wash 50-70 third instar larvae as described in Step 1. Place the larvae in a 1.5 ml microfuge tube containing 500 μl of RNAse free 1X PBS.
- Dounce the larvae using a polypropylene pestle (USA Scientific), with 6-7 slow powerful strokes.
- Centrifuge the solution at 16,000 (x) g for 5-10 seconds (until the larval cuticles settle down to the bottom of the microfuge tube]. Discard the supernatant. The pellet should primarily consist of larval cuticle to which the PNS, including da neurons, is tightly adhered.
- Wash the cuticle pellet 2-3 times in 1X PBS and resuspend the pellet in 500 μl 1X PBS. Spin the solution at 16,000 (x) g for 1 minute to form a compact pellet. Aspirate the supernatant completely using a fine Pasteur pipette.
- Carefully remove the pellet from the microfuge tube using a clean spatula, and wick off the excess PBS using a clean Kimwipe tissue. [Note: It is important to keep the pellet as compact as possible to increase the cell yield]. Place the compact cuticle pellet on a plastic mold containing a thin layer of OCT (1mm approx).
- Gently spread out the pellet in a thin circular area. Fill the mold with OCT, and freeze the tissue as described in Step 4.
- Using a cryostat, cut frozen sections at 5-8 μm thickness on plain, labeled, uncharged, RNAse free glass microscope slides. [Critical Step: Position the tissue sections near the center of the slide.]
- Store the slides either directly on dry ice or at -80°C in a clean slide-box until ready for microdissection. Perform LCM preferably within a week after sectioning the tissue.
PAUSE POINT
2. Dehydration and Cuticle Removal from Frozen Larval Sections
[Critical Step: All solutions for dehydration must be prepared fresh before each LCM session. Complete dehydration of frozen larval tissue sections is essential for achieving optimal microdissection efficiency]
- Remove the slides containing the frozen larval tissue sections from the -80°C freezer and place them on dry ice.
- Remove a single slide from dry ice and immediately place it directly into a 50 ml conical tube filled with 70% ethanol fixative, followed by a short rinse in RNAse free ddH2O according to the recommended times presented in Table 1.
- The presence of larval cuticle in the frozen sections may impair the capture efficiency by preventing efficient LCM cap-cell contact which may lead to non-specific capture. If necessary, conduct the following steps (4-5) to clear the cuticle from tissue sections.
- Gently pipette 50 μl of 2.5% trypsin directly onto the tissue sections and incubate for 5-30 seconds at room temperature. [Critical Step: This step may require optimization. The incubation time depends upon the tissue to be microdissected and section thickness. Longer incubation may clear everything including the cells of interest, whereas a short incubation may be insufficient to remove the cuticle]
- Rinse the sections briefly in a 50 ml conical tube containing RNAse-free ddH2O to remove trypsin, and loosely adhered cuticle fragments.
- Dip the slides sequentially in each of the remaining ethanol and xylene solutions for the recommended times (Table 1) to complete the dehydration process.
- Following the dehydration gradient, the slides are briefly dried under a mild air stream for 60-120 seconds at room temperature prior to performing LCM. [Caution: Dry the xylene completely from the tissue sections prior to performing LCM. Xylene is known to dissolve the LCM cap polymer resulting in microdissection failure].
| 70% Ethanol (Fixative) | 3-10 Seconds |
| ddH2O | 5-10 Seconds |
| 2.5% Trypsin Incubation | 5-30 Seconds |
| ddH2O | 5-10 Seconds |
| 70% Ethanol | 60 Seconds |
| 95% Ethanol | 60 Seconds |
| 100% Ethanol | 120 Seconds |
| 100% Ethanol | 120 Seconds |
| 100% Xylene | 120 Seconds |
| 100% Xylene | 120 Seconds |
| Air dry in a mild air stream | 60-120 Seconds |
Table 1: Recommended procedure for LCM dehydration of frozen larval tissue sections.
3. Laser Capture Microdissection
PixCell IIe LCM instrument equipped with Fluor 300 epifluorescence optics optimized for EGFP was used for performing the LCM.
[Critical Step: If possible, perform the microdissection in a humidity-controlled room to prevent any reduction in microdissection efficiency due to increased humidity. Room humidity is a critical factor affecting the microdissection efficiency. Low room humidity can cause an increased static-cling resulting in non-specific capture, while high room humidity can result in low microdissection efficiency. We achieved optimal LCM efficiency between 25-50% relative humidity conditions.]
- Turn on the power for the microscope and laser control box.
- Load the CapSure cap holder assembly with HS LCM caps (Molecular Devices): Two cartridges of LCM caps may be loaded at one time.
- Open the Molecular Devices software and enter the experiment details including slide number and Cap lot number.
- Load a new HS LCM cap from the cartridge onto the PixCell IIe LCM instrument and position it correctly with respect to the joystick to ensure proper positioning of the cap in relation to the capture area.
- Place the microscope slide containing freshly dehydrated larval tissue sections on the microscope stage for microdissection.
- Locate the fluorescently labeled cells using the oculars or the computer screen. Due to the high specificity of ppk-GAL4,UAS-mCD8::GFP transgenic reporter line, the class IV da neurons can be easily identified by GFP fluorescence.
- Subject the tissue to LCM using CapSure HS LCM caps [for a detailed set of instructions for setting the instrument, focusing the laser and performing LCM see reference22].
- Adjust the “Power” and “Duration” laser pulse parameters to achieve a precise melted polymer spot, whose size corresponds to the selected laser spot size. Adjust the settings to customize the size of melted polymer area. The following parameters were used for microdissecting single class IV da neurons: Spot Diameter 7.5 μm, laser strength 30-50 mW, laser time and 2-4 ms and 1-2 shots per cell were used. [Critical Step: the laser parameters required for a proper spot may vary with tissue thickness and room humidity conditions. Adjust the “Power” and “Duration” settings to achieve the required melted polymer spot size for microdissecting single or multiple cells.]
- To maximize specificity, microdissect cells with minimal overlap and a strong fluorescence. Each cap is capable of capturing numerous cell bodies. [Critical Step: To avoid RNA degradation, limit the total analysis time including dehydration and microdissection to less than 45 minutes]
- Once all the cells of interest have been captured, lift the HS LCM cap with the microdissected cells and placed it on a clean slide to confirm the presence of the captured cells and visually inspect the cap for presence of unwanted cells or debris.
4. RNA isolation from LCM derived cells
- Attach the cap containing the microdissected cells to an ExtracSure (Molecular Devices) device. Add 12μl of RNA extraction buffer (PicoPure, Molecular Devices) to the cap surface containing the cells and connect an inverted thin-walled reaction tube (GeneAmp, Applied Biosystems) to the ExtracSure device. Place the assembly on an alignment tray (Molecular Devices) and cover it with an incubation block (Molecular Devices) pre-heated to 42°C inside an incubator.
- Following a 30 minute incubation, centrifuge the tubes containing the extracts at 800 (x) g for 2 minutes. Close the tubes and store the cell extracts at -80°C until ready for RNA purification.
PAUSE POINT
- Extract and column purify the RNA according to the PicoPure (Molecular Devices) RNA extraction kit instructions. DNAse treatment is optional, and can be performed on column during the RNA purification as necessary. If required, multiple LCM samples can be pooled together during purification in a single column to increase the final RNA yield and can be eluted in a small volume (11-30 μl) of elution buffer (PicoPure, Molecular Devices) and stored at -80°C until ready for use. If desired a 1 μl aliquot may be used to assess total RNA quality on a Bioanalyzer 2100 (Agilent Technologies, Inc.).
Representative Results
Laser capture microdissection (LCM) was used to isolate single, class-specific or multiple Drosophila da neurons from third instar larvae (Figure 1). LCM allows for a high degree of precision and specificity in the isolation of Drosophila da neuron cell bodies (Figure 2a-c). Moreover, the total RNA purified from these isolated da neurons was found to be of excellent quality as indicated by the presence of sharp 5.8S, 18S and 28S ribosomal RNA peaks when analyzed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.) (Figure 2d).
To assess the neuronal-specific enrichment of our isolated cells we performed real-time quantitative reverse transcription PCR (qRT-PCR) using the neuronal gene-specific marker, elav. For the qRT-PCR analyses, we calculated the relative levels of elav expression in the LCM microdissected da neurons and normalized this expression to that of our endogenous control (rp49) and relative to total RNA isolated from whole larvae using the ΔΔCt method23. These analyses revealed over 2.5 fold enrichment in the relative levels of elav in LCM micro-dissected da neuron samples as compared to whole animals (Figure 3).
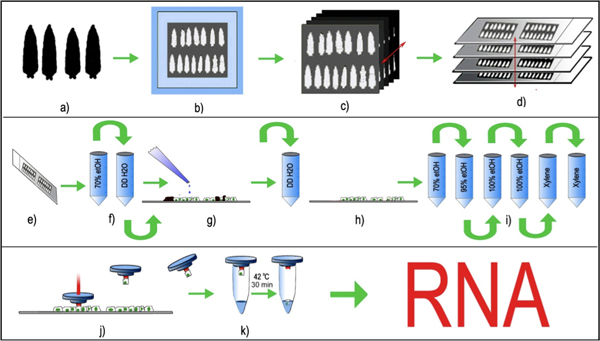
Figure 1: LCM of Drosophila peripheral neurons. (a) Age matched third instar larvae are selected, washed and (b) embedded in a cryomold with OCT and frozen at -80°C, (c) 8μm serial tissue sections are created using a standard cryostat and (d) placed uniformly on clean glass slides. The glass slides with the affixed tissue sections are stored at -80°C prior to LCM processing. (e,f) Immediately preceding LCM, the tissue sections are thawed and briefly fixed in 70% ethanol followed by brief rinse in ddH2O. (g) The slides are briefly incubated with trypsin and rinsed with ddH2O to remove cuticles (black) from the larval sections. (h,i) tissue sections without larval cuticle are then further dehydrated in an ethanol gradient and finally cleared in xylene. (j) The LCM cap with a thermolabile polymer is placed on the tissue section and the laser is pulsed on the selected fluorescent cell body. The laser pulse melts the polymer and engulfs the selected cell body. The polymer cap, along with the captured cell body is lifted, and (k) RNA extraction buffer is added to the cell. The captured cell, along with the RNA extraction buffer is incubated at 42°C for 30 minutes and can be either stored at -80°C, or directly processed for RNA purification.
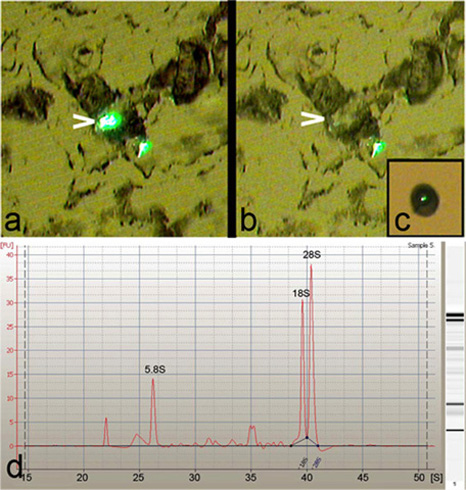
Figure 2: LCM facilitates high precision capture of da neurons. (a) Representative image of a dehydrated and trypsin treated 8 micron tissue section prior to performing LCM, showing two class-IV da neurons labeled with GFP by the ppk-GAL4,UASmCD8::GFP reporter strain. Note, one of the neurons is highlighted (arrowhead) for capture. (b) Cell body of the highlighted neuron (arrowhead) is cleanly micro-dissected with high specificity from the tissue section. (c) End-on view of a single class-IV da neuron captured on the LCM cap. (d) Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.) electropherogram of total RNA isolated from LCM-derived da neurons, showing excellent RNA quality as indicated by the presence of sharp 5.8S, 18S, and 28S rRNA peaks.
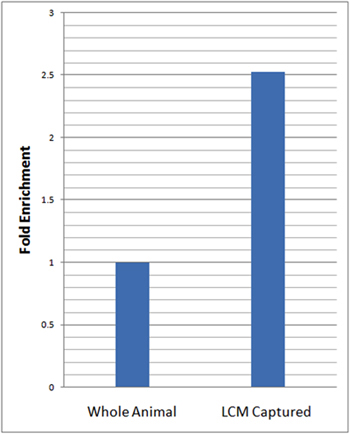
Figure 3: qRT-PCR reveals substantial enrichment of neuronal marker gene expression in LCM captured da neurons relative to whole animal. qRT-PCR analyses of neuron-specific marker gene expression (elav) in LCM captured da neurons (21-7-GAL4,UAS-mCD8::GFP) and whole animals in age matched third instar larvae was performed in triplicate. Relative levels of elav expression in LCM captured da neurons were normalized to the endogenous control (rp49) and relative to whole larvae using the ΔΔCt method23. A 2.5 fold enrichment in the relative levels of elav in LCM captured da neuron samples was observed relative to whole animals.
Troubleshooting
Problem: RNA degraded, or of low quality.
Ensure RNAse free condition throughout experiment. Try reducing amount of time spent on LCM per slide to 30 minutes. Keep the slides and samples on dry ice except when performing LCM. Avoid thawing the tissue sections once they are cut.
Problem: Most cells are lost from the slide during trypsin treatment (step 17-18).
Try reducing the time of trypsin treatment. Try reducing the trypsin concentration.
Problem: Presence of cuticle and other non-specific tissue debris on the cap polymer.
Try removing the tissue debris by blotting the polymer surface with the tacky side of an adhesive note22.
Problem: Low LCM efficiency.
Try increasing the incubation time in 100% ethanol and xylenes. Reduce room humidity using dehumidifiers.
