1. Cloning and Construct Design
- Clone the codon-optimized sequence (for E. coli) of C. difficile PPEP-1 without the signal peptide [amino acids 27-220, named hereafter recombinant PPEP-1 (rPPEP-1)11] into the pET28a vector using NdeI and XhoI restriction sites (Figure 1) with a stop codon at the 3'-end (resulting vector pET28a-NHis-rPPEP-1). This produces a N-terminally 6xHis-tagged protein (NHis-rPPEP-1) with a thrombin cleavage site allowing removing the tag during purification (Figure 1). The plasmid contains a kanamycin resistance cassette for selection. The primers used for cloning are described elsewhere14.
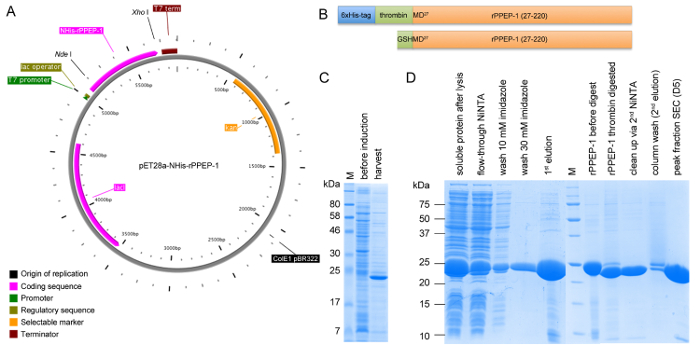
Figure 1: Schematic representation of construct pET28a-NHis-rPPEP-1 and SDS-PAGE analysis of expression and all purification steps. (A) Vector map of NHis-rPPEP-1 cloned into pET28a vector using NdeI/XhoI created with PlasMapper. (B) Schematic representation of the NHis-rPPEP-1 construct (upper panel) and the final construct after thrombin-cleavage of the 6xHis-tag with the resulting additional GSHM-sequence at the N-terminus (lower panel). SDS-PAGE analysis (C) of the expression in BL21 (DE3) Star at 37 °C for 4 hr and (D) of samples from all purification steps (M: molecular weight marker). Please click here to view a larger version of this figure.
2. Expression and Purification of rPPEP-1
- Expression of NHis-rPPEP-1
- Make up and autoclave LB (lysogeny broth) medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl, adjust to pH 7.5 with NaOH). Supplement with kanamycin sulfate (50 µg/ml) just before use (LB/Kan medium).
- Inoculate a 200 ml overnight culture from freshly transformed E. coli in LB/Kan medium. Grow overnight at 37 °C with shaking at 220 rpm.
- On the next morning, check the OD600 (optical density at 600 nm wavelength) of the overnight culture. Inoculate two 2.8 L baffled flasks containing 1 L LB/Kan medium each with the overnight culture to an OD600 of 0.1. Supplement with three drops of aqueous-silicone emulsion to prevent excessive foam formation. Grow cells at 37 °C shaking at 180 rpm until the OD600 reaches 0.6.
- Take a pre-induction sample for SDS-PAGE analysis (equivalent of 1 ml from a culture at OD600 = 1); add IPTG to 0.5 mM final concentration to induce expression of NHis-rPPEP-1. Continue growing at 37 °C/180 rpm for 4 hr.
- Determine the OD600 in a 10x dilution and take a harvest sample (equivalent of 1 ml from a culture at OD600 = 1).
- Collect cells by centrifugation for 20 min at 7,000 x g and 4 °C. To remove residual LB medium resuspend cell pellets from 1 L of culture in 40 ml TBS buffer (Tris-buffered saline: 20 mM Tris-HCl, pH 7.5, 200 mM NaCl) and transfer to a 50 ml centrifuge tube. Collect cells by centrifugation for 10 min at 10,000 x g and 4 °C and store at -80 °C until use. Analyze expression (total lysates and soluble fractions) via SDS-PAGE18.
- Purification of untagged rPPEP-1
- Take 50 µl samples of each purification step for SDS-PAGE analysis. Resuspend the cell pellet from 1 L of culture in TBS buffer supplemented with 10 µg/ml DNaseI. Use 5 ml of TBS/DNaseI per g of cells.
- Lyse the cells by sonication on ice/water using 30% amplitude for 15 min (2 sec pulses with 2 sec pause). Remove debris by centrifugation for 10 min at 10,000 x g and 4 °C and transfer supernatant to an ultracentrifuge tube. Clear lysate in an ultracentrifuge for 30 min at 165,000 x g and 4 °C.
- Work at 4-6 °C. Using a peristaltic pump or chromatography system equilibrate 2 ml of nickel-nitrilotriacetic acid (NiNTA) resin in a glass column with TBS buffer supplemented with 10 mM imidazole pH 7.5. Alternatively, use gravity flow.
- Adjust the cleared lysate with 1 M imidazole pH 7.5 to a final concentration of 10 mM. Apply the lysate to the column and wash stepwise with TBS buffer supplemented with 10 mM and 30 mM imidazole, respectively, until the UV absorption at 280 nm has reached the baseline.
- Elute the protein with TBS buffer plus 250 mM imidazole. Re-equilibrate the column to TBS supplemented with 10 mM imidazole and store overnight.
- Determine the protein concentration either at 280 nm by using the extinction coefficient of 25,900 M-1 cm-1 or by any other method (e.g. Bradford method19). Add 2 units of thrombin per mg of protein and dialyze the protein solution overnight at 4 °C against a 50x volume of TBS (50x of the NiNTA elution volume).
NOTE: Take the correct blank for determination of protein concentration, as imidazole absorbs strongly at 280 nm. - Pass the protein solution over the equilibrated NiNTA resin to remove uncleaved protein. Next, apply the same volume of TBS supplemented with 10 mM imidazole to the column to recover all cleaved protein. To clean the column, elute all remaining protein with 250 mM imidazole. Analyze samples via SDS-PAGE (Figure 1).
- Concentrate the protein solution to 4 ml in 10 min intervals at 4,000 x g and 4 °C using a centrifugal ultrafiltration unit. Mix the concentrating protein after each interval to prevent precipitation and aggregation. At this step occasionally some precipitation is observed for rPPEP-1 despite the mixing procedure.
- Apply the concentrated protein to a pre-equilibrated size exclusion chromatography column (in TBS buffer) at 4-6 °C. Elute the column with TBS buffer, collect 1 ml fractions and subject 5 µl of every second fraction to SDS-PAGE analysis. rPPEP-1 elutes in a single peak corresponding to a monomer (Figure 2). Occasionally a minor peak at larger molecular weight is observed (fronting peak), that corresponds to a dimer of the protein. The yield should be around 50 mg of pure protein per L of culture. Analyze all samples via SDS-PAGE18 (Figure 2).
- Take 50 µl samples of each purification step for SDS-PAGE analysis. Resuspend the cell pellet from 1 L of culture in TBS buffer supplemented with 10 µg/ml DNaseI. Use 5 ml of TBS/DNaseI per g of cells.
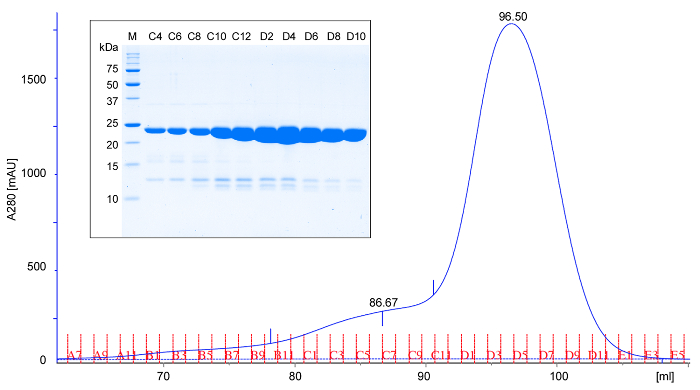
Figure 2: Representative size exclusion chromatography and SDS-PAGE analysis of rPPEP-1. Size exclusion chromatogram (A280; absorbance at 280 nm) of purified untagged rPPEP-1 using a (16/600) column in Tris-HCl, pH 7.5, 200 mM NaCl at 6 °C. Based on the elution volume, rPPEP-1 migrates as expected for a 22 kDa protein, suggesting that it is predominantly monomeric. Rarely a minor fronting peak appears that corresponds to a dimer. (inset) SDS-PAGE analysis of the fractions from size exclusion chromatography (M; molecular weight marker). Every second fraction is applied. The faint bands below the main rPPEP-1 band correspond to occasionally occurring minor impurities. Please click here to view a larger version of this figure.
3. Crystallization and Crystal Optimization Using Microseeding
NOTE: rPPEP-1 crystallizes from conditions that constantly produce highly intergrown crystals not suitable for X-ray diffraction analysis (Figure 3). Therefore, an optimization strategy (Figure 4) was developed to obtain high quality crystals (Figure 5).
- Initial screening of rPPEP-1 using commercial screens
NOTE: Carry out crystallization trials in the sitting drop format using standard commercially available screens and a crystallization robot.- Concentrate the purified protein to 12 mg/ml using a centrifugal ultrafiltration device in 5 minute intervals at 4,000 x g and 4 °C. Mix the concentrating protein after each interval to prevent precipitation and aggregation. Determine the protein concentration either at 280 nm by using the extinction coefficient of 25,900 M-1 cm-1 or by any other method (e.g. Bradford method). Equilibrate the protein to 20 °C. Clear away all particles and dust by centrifugation for 10 min at 16,000 x g and 20 °C.
- Either use already pre-filled crystallization plates sealed and stored at 4 °C or fill the reservoir wells of the plates with 70 µl of every crystallization condition. Equilibrate all crystallization plates to 20 °C. Work quickly, as the small volumes quickly dry out. Use a humidity chamber around the dock of the robot, if possible.
NOTE: Use the following screens as a standard procedure: SaltRx, Index, PEG/Ion, Crystal, Wizard, PACT++, JCSG++. - Set up the screen by pipetting protein and reservoir into subwells 2-4. Drop volume is 300 nl and the ratios (protein:reservoir) are 200:100 (subwell 2), 150:150 (subwell 3) and 100:200 (subwell 4) (in nl). Immediately seal the plate and place in a chamber at 20 °C.
- Inspect trays immediately after set-up, and then inspect every day during the first week followed by weekly inspection.
- Co-crystallization of rPPEP-1 with ligands
- For co-crystallization of substrate peptide-rPPEP-1 complexes mix rPPEP-1 at 24 mg/ml in a 1:1 ratio (v/v) with a 7-fold molar excess of peptide solution (Ac-EVNPPVPD-NH2) lyophilized powder solubilized in TBS buffer), which will give a final concentration of 12 mg/ml r-PPEP-1 protein and a 7-fold molar excess of peptide over PPEP-1. Incubate for 30 min at 20 °C and clear away all particles and dust by centrifugation for 10 min at 16,000 x g and 20 °C. Proceed with crystallization using the microseeding procedure as described for the unbound r-PPEP-1 protein.
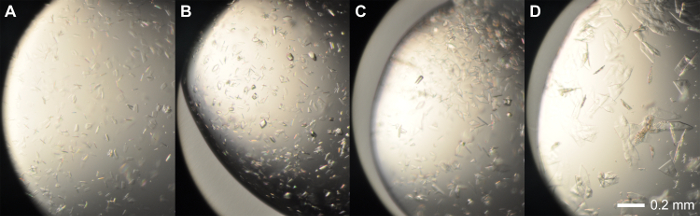
Figure 3: Representative crystals from initial screens. Intergrown crystals from rPPEP-1 at 12 mg/ml grown in condition. (A) Crystal screen I/38 (1.4 M sodium citrate tribasic dehydrate, 0.1 M HEPES sodium pH 7.5; 200 nl:100 nl). (B) SaltRx screen/52 (2.4 M ammonium phosphate dibasic, 0.1 M Tris pH 8.5; 100 nl:200 nl) and (C) (200 nl:100 nl). (D) SaltRx screen/96 (60% v/v Tacsimate pH 7.0, 0.1 M BIS-TRIS propane pH 7.0; 200 nl:100 nl). The Scale bar = 0.2 mm. Volume ratios are always protein:reservoir. Please click here to view a larger version of this figure.
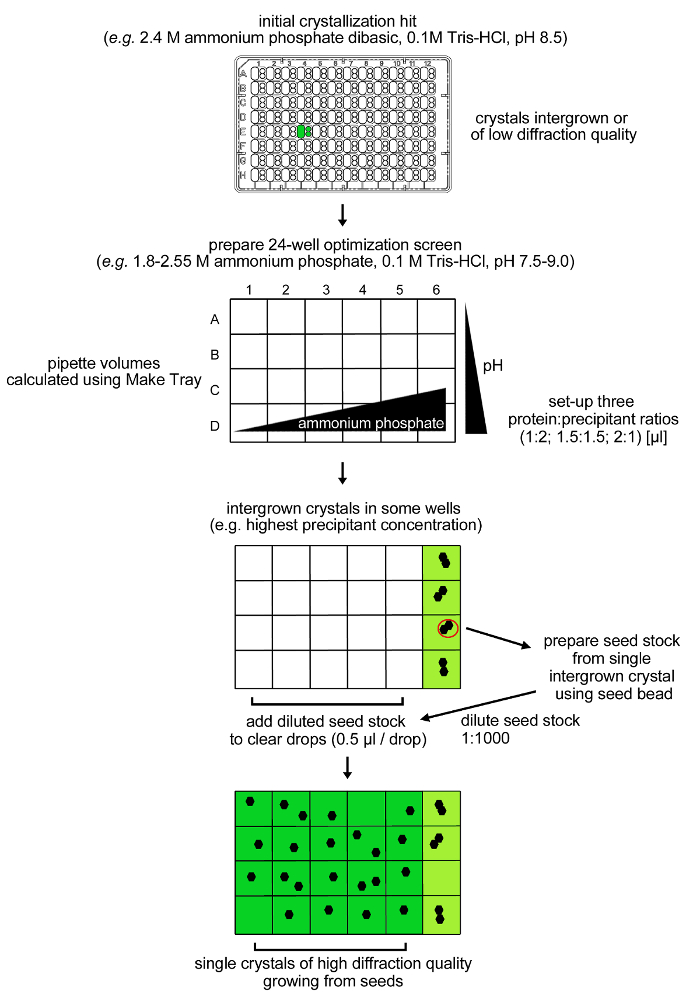
Figure 4: Optimization procedure for rPPEP-1 crystallization. Initial crystals from rPPEP-1 at 12 mg/ml of low diffraction quality and with multiple lattices (intergrown) were reproduced in a 24-condition optimization screen. Again, only intergrown crystals were observed in conditions containing 2.55 M ammonium phosphate dibasic. A seed stock was prepared from a single intergrown crystal and diluted 1:1,000 into the same condition (microseeding). A volume of 0.5 µl of the diluted seed stock was added into the remaining clear drops and single crystals grew in almost all conditions. Please click here to view a larger version of this figure.
- Crystal optimization using microseeding
NOTE: Highly intergrown crystals of rPPEP-1 appear after two days in a condition containing 2.4 M ammonium phosphate dibasic, 0.1 M Tris-HCl, pH 8.5 (SaltRx screen, condition E4, all three subwells) (Figure 3). An optimization procedure using a grid screen around the initial condition combined with microseeding was applied (Figure 4).- Prepare a grid screen (Figure 4) comprising 24 conditions with 1.8 – 2.55 M ammonium phosphate dibasic (in steps of 0.15 M) and 0.1 M Tris-HCl pH 7.5-9.0 (in steps of 0.5 pH-units) from appropriate stock solutions (4 M ammonium phosphate and 1 M Tris buffers).
NOTE: Use the Make Tray applet (http://hamptonresearch.com/make_tray.aspx) to calculate the volumes and the pipetting scheme to obtain 2 ml of every condition that allows to perform 10 optimization screens. The 4 M ammonium phosphate stock solution is difficult to prepare. Heat the solution during stirring to completely dissolve the powder in water. - Pipette 200 µl of every grid screen solution into the wells of a 24-well plate and equilibrate at 20 °C.
- Manually set up the crystallization plate. The drop volume is 3 µl and the ratios (protein:reservoir) are 2:1, 1.5:1.5 and 1:2 (in µl). Here, use a positive displacement pipette to avoid air bubble formation. Immediately seal the plate and place in a chamber at 20 °C. Avoid air bubble formation.
NOTE: After one to four days highly intergrown crystals appear in the four conditions containing 2.55 M ammonium phosphate dibasic and 0.1 M Tris-HCl pH 7.5 – 9.0 (Figure 4). No crystals are formed in the remaining 20 conditions with ammonium phosphate concentrations below 2.55 M. The microseeding procedure is used to obtain single crystals of rPPEP-1 in these conditions. - Prepare a microseed stock by harvesting a single intergrown crystal from one of the two conditions with 2.55 M ammonium phosphate dibasic and 0.1 M Tris-HCl pH 8.0 or 8.5. The crystals might be attached to the plastic surface. Careful deformation of the surrounding plastic with an acupuncture needle helps to detach the crystals.
- Transfer 50 µl of the respective mother liquor into a 1.5 ml tube containing a small highly polished glass bead (beads-for-seeds). Using a mounted nylon loop transfer the crystal into 1 µl of the mother liquor placed onto a glass cover slide.
- Transfer the liquid containing the crystal into the tube and vortex at high speed for 30 sec. Make a 1:1,000 dilution of the seed stock into a new 1.5 ml tube containing the same freshly prepared condition and vortex thoroughly for 5 seconds.
NOTE: Seeds stocks can be stored at -80 °C for later use.
- Remove the seal of the plate covering the 20 conditions with clear drops and pipette 0.5 µl of the seed stock (1:1,000 dilution) into the wells. Seal the plate and place in a chamber at 20 °C. Single crystals of high diffraction quality appear in 2-7 days (Figure 5).
- Prepare a grid screen (Figure 4) comprising 24 conditions with 1.8 – 2.55 M ammonium phosphate dibasic (in steps of 0.15 M) and 0.1 M Tris-HCl pH 7.5-9.0 (in steps of 0.5 pH-units) from appropriate stock solutions (4 M ammonium phosphate and 1 M Tris buffers).
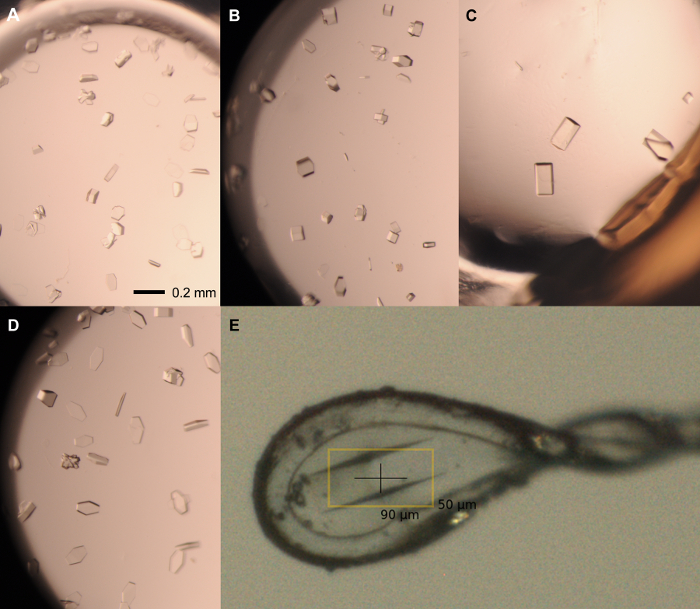
Figure 5: Representative crystals from optimization screen. Single crystals from rPPEP-1 at 12 mg/ml seeded with 1:1,000-dilution seed stock grown in the following conditions: (A) 2.1 M ammonium phosphate dibasic, 0.1 M Tris pH 7.5; 1.5 µl:1.5 µl; (B) 2.1 M ammonium phosphate dibasic, 0.1 M Tris pH 7.5; 2 µl:1 µl; (C) 2.25 M ammonium phosphate dibasic, 0.1 M Tris pH 8; 2 µl:1 µl; (D) 2.1 M ammonium phosphate dibasic, 0.1 M Tris pH 8; 1 µl:2 µl. (E) Mounted crystal in 0.1-0.2 µm nylon loop, grown in 2.1 M ammonium phosphate dibasic, 0.1 M Tris pH 8 (2 µl:1 µl) and cryo-protected in 2.1 M ammonium phosphate dibasic, 0.1 M Tris pH 8, 20% glycerol. The Scale bar = 0.2 mm in (A-D). Volume ration are always protein:reservoir. Please click here to view a larger version of this figure.
4. Crystal Mounting and Data Collection
NOTE: To obtain the best quality of diffraction data crystals should be mounted at the peak of their quality and size. Crystals can be stored in liquid nitrogen until they are subjected to X-ray diffraction analysis at 100 K. Therefore, the condition from which they originate must be adjusted to cryo-conditions. rPPEP-1 crystals can be cryo-protected by addition of either 20% glycerol or 30% sucrose (replacement of water in the condition by the cryo-protectant).
- Crystal mounting
NOTE: All crystal manipulation steps should be performed under the stereomicroscope.- Choose the optimal size of nylon loop for the maximal length of the chosen crystals. The typical longest axis of rPPEP-1 crystals is about 100-200 µm (Figure 5). Prepare a cover slide and the appropriate cryo-condition (e.g. 2.1 M ammonium phosphate dibasic, 0.1 M Tris, pH 8.0, 20% glycerol).
- Fill the foam dewars with liquid nitrogen, load the vial clamp with a vial and pre-cool it in the liquid nitrogen-filled 800 ml foam dewar. Place a cryo sleeve and a cryo cane holder marked with a suitable identifier in liquid nitrogen-filled 2 L foam dewar. Load the magnetic wand with a mounted nylon loop.
NOTE: Wear protective clothing (eyeshield/glasses, gloves) when working with liquid nitrogen. Warm objects plunged into liquid nitrogen may produce spills.
- Fill the foam dewars with liquid nitrogen, load the vial clamp with a vial and pre-cool it in the liquid nitrogen-filled 800 ml foam dewar. Place a cryo sleeve and a cryo cane holder marked with a suitable identifier in liquid nitrogen-filled 2 L foam dewar. Load the magnetic wand with a mounted nylon loop.
- Cut open the sealing tape with a sharp scalpel and remove it with the forceps. Pipette 1 µl of the cryo-condition onto the cover slide (or alternatively in an empty well on the same plate) and remove the crystal from the drop by fishing it with the mounted nylon loop (Figure 5). Attached crystals can be easily detached from ground by deforming the surrounding plastic with an acupuncture needle.
- Quickly transfer the crystal to the drop of cryo-condition and let it equilibrate for 1 second. Fish the crystal out as quickly as possible and plunge-freeze in liquid nitrogen.
- When the liquid nitrogen around the mounted loop stops boiling, place the loop in the vial. Place the vial on the cryo cane holder and when loaded with 6 vials place a cryo sleeve around the holder. Store the crystals in a tank filled with liquid nitrogen until use.
- Choose the optimal size of nylon loop for the maximal length of the chosen crystals. The typical longest axis of rPPEP-1 crystals is about 100-200 µm (Figure 5). Prepare a cover slide and the appropriate cryo-condition (e.g. 2.1 M ammonium phosphate dibasic, 0.1 M Tris, pH 8.0, 20% glycerol).
- Data collection
NOTE: Data collection can be carried out at the home diffractometer, if available, or at a synchrotron beamline. For rPPEP-1 data were collected at the beamline X06DA of the Swiss Light Source, Paul-Scherrer-Institute, Villigen, Switzerland using a hybrid photon counting detector. The original data and all files used in structure determination can be provided upon request.- Set up the wavelength of the beam to 1.282 Å (9,667 keV), which is the characteristic x-ray absorption edge energy (peak) of the element zinc. rPPEP-1 is a metalloprotease that contains a single zinc per molecule in the active site.
- Collect data at 100 K in the inverse-beam mode in 10° wedges for a total of 270° in each direction. The exposure time is 0.1 sec with 0.1° rotation per image. Set the transmission to 14% (0.14).
- To collect a native high-resolution dataset from a second crystal originating from the same crystallization condition set up the wavelength of the beam to 1.00 Å (12,398 keV). Collect data at 100 K. The exposure time is 0.1 sec with 0.1° rotation per image. Set the transmission to 70% (0.7).
5. Structure Determination via Zinc-SAD
NOTE: In order to determine the structure of rPPEP-1 via zinc-SAD some basic crystallographic knowledge is needed as well as the software packages XDS20, Phenix21 and the program Coot22. For visualization of structures the program PyMOL23 or Chimera24 is needed. Data collected at the wavelength corresponding to the peak at the absorption edge of the element zinc can be used for single-wavelength anomalous dispersion (SAD)25 to obtain phase information that can be extended for all protein atoms.
- Data processing
- Process the two peak datasets (normal and inverse) using the software XDS (alternatively iMosflm or HKL3000) in space group P212121 (space group 19) separating the Friedel's mates (anomalous data). The unit cell parameters should be around a, b, c (Å) = 43.17, 71.68, 117.70 and α=β=γ (°) = 90. This gives two HKL-files (reflection files).
- Inspect the file CORRECT.LP. Use data up to the resolution in which the CC1/2 is at least 50%. Scale together both datasets/reflection files (HKL-files) using XSCALE. Inspect the file XSCALE.LP. Check how far the anomalous signal extends (SigAno) and note the resolution with an anomalous correlation (Anomal Corr) of about 30%, which is 2 Å in the case of the data used here collected to 1.67 Å. This is the resolution cut-off for the anomalous signal used in Phenix AutoSol.
- Convert the (scaled) HKL-file into a CCP4-format reflection file (named, for example, peak_anom.mtz) using XDSCONV creating an Rfree subset of 5% and keeping the anomalous data (FRIEDEL'S_LAW=FALSE). Check the mtz-file for consistency with the program mtzdmp inspecting the unit cell parameters, the space group and the existence of the Rfree subset (label FreeRflag) and the anomalous data (labels DANO/SIGDANO). Prepare also an additional mtz-file with XDSCONV without extracting the anomalous data (FRIEDEL'S_LAW=TRUE; named, for example, peak_native.mtz) for refinement at a later stage.
- Substructure solution (phase determination)
- Run Phenix AutoSol using the reflection file peak_anom.mtz. Select SAD/MAD peak as data type and choose 2 zinc sites (as there are two molecules per asymmetric unit). Choose either the more exact experimental values for the f'/f'' parameters (determined in a fluorescence scan at the beamline) or the theotetical values f' = -8.245 and f'' = 3.887. Also load the FASTA file containing the amino acid sequence of the crystallized protein.
- Set the resolution limit to the resolution with an anomalous correlation (Anomal Corr) of about 30% (determined in 5.1.2), in this case 2 Å and select the "autobuild model" option. Using the phases of the two zinc sites found by Phenix HySS (part of the Phenix AutoSol pipeline) the phases for the whole protein could be deduced and the model built (by Phenix RESOLVE) into the electron density. The best model is called "overall_best.pdb".
- Model building, refinement and validation
- Select the option "autobuild model" to build most of the rPPEP-1 model automatically. Inspect the electron density at 1.0 σ contour level using the program Coot (Figure 6). It should be connective and surrounding the atoms of the model. Ideally also some water molecules should be built into the model (at a resolution better than 2.5 Å). Bulk water (space between the molecules) should contain no density.
- Check if the whole model is complete (all amino acids built into the electron density). If not, build them manually using the tools provided by Coot. Refine the structure by running iterative rounds of Phenix Refine with 5 refinement rounds each using the overall_best.pdb model file, the peak_native.mtz reflection file and the FASTA sequence file; and manual model building in Coot.
- Validate the quality of the structural model with the respective tools in Coot.
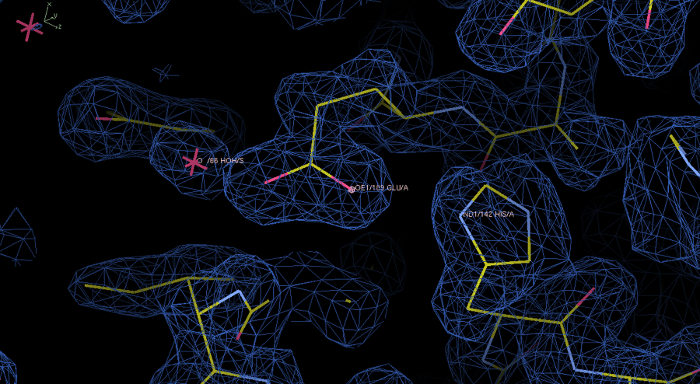
Figure 6: Experimental electron density map and model of rPPEP-1 after the Phenix Autosol run. Electron density in blue at a contour level of 1.0 σ displayed in the program Coot. In this initial map the electron density is nicely resolved and the model build into the electron density. The zoom in shows the residues His142 and Glu189, as well as a water molecule. Please click here to view a larger version of this figure.
6. Structure Determination to High Resolution via Molecular Replacement
NOTE: In order to obtain high-resolution structural information about rPPEP-1 a native dataset is collected. Then a molecular replacement procedure using the software Phaser26,27 (within the Phenix software package) is employed using the structure determined via zinc-SAD as a model. This procedure can be also used later when solving structures of rPPEP-1 complexed with small molecules.
- To obtain a crystal structure with higher resolution (in this case up to 1.4 Å) process the native dataset using the software XDS (alternatively iMosflm or HKL3000) in space group P212121 (space group 19). The unit cell parameters should be around a, b, c (Å) = 43.17, 71.77, 117.80 and α=β=γ (°) = 90. This gives one HKL-files (reflection file).
- Inspect the file CORRECT.LP. Use data up to the resolution in which the CC1/2 is at least 50%. Convert the HKL-file into a CCP4-format reflection file (named, for example, native.mtz) using XDSCONV creating an Rfree subset of 5%. Check the mtz-file for consistency with the program mtzdmp inspecting the unit cell parameters, the space group and the existence of the Rfree subset (label FreeRflag).
- Prepare the PDB-file containing the model from overall_best.pdb determined earlier and remove all water molecules and all ligands (i.e. the zinc atom). Also load the FASTA file containing the amino acid sequence of the crystallized protein. Run Phaser in Phenix using the reflection file native.mtz. Search for two molecules per asymmetric unit.
- After successful structure solution (TFZ-score larger than 8; here 10.2) inspect the model (named native_phaser.1.pdb) and the electron density map in Coot. Build and refine the structure by running iterative rounds of Phenix Refine with 5 refinement rounds each using the native_phaser.1.pdb model file, the native.mtz reflection file and the FASTA sequence file; and manual model building in Coot.
- Validate the quality of the structural model with the respective tools in Coot.
X-ray crystallography is still the fastest and most accurate method to determine three-dimensional near-atomic resolution structures of proteins28. However, it requires the growth of well-ordered single crystals. These are often difficult to get and the crystalline state is artificial. However, a comparison of protein structures determined by X-ray crystallography with those determined by other methods, especially NMR, shows generally a very good agreement. In the case of PPEP-1, an NMR structure published recently29 shows excellent agreement with our crystal structure14, including the mobility of the S-loop.
This protocol describes the production and purification of N-terminally His-tagged rPPEP-1 protein for structural studies and the crystallization and structure determination of untagged rPPEP-1. Single crystals were difficult to grow in this case and required a special microseeding procedure. In the following section we will discuss the results for PPEP-1 and indicate how the protocol could be adapted for the production and crystallization of any other protein.
Variations on construct design and expression
rPPEP-1 is expressed as an N-terminally His-tagged variant (Figure 1A), as for crystallization it is preferable to remove the His-tag by thrombin digest due to its possible impact on crystallization success and protein structure30. For other proteins it may be advisable to additionally test a C-terminally His-tagged version (e.g. cloned using the same restriction sites, the vector pET22b and a reverse primer without stop codon at the 3' end) or to leave the N-terminal His-tag uncleaved, as in some cases a tag may help during crystallization. For rPPEP-1 the yield and stability of a C-terminally His-tagged construct was inferior. Additionally, an initial testing for best soluble protein expression should be included in the following E. coli strains: BL21 (DE3), BL21 (DE3) pLysS or Lemo21 (DE3), BL21 (DE3) codon plus RIPL or Rosetta 2 (DE3), BL21 (DE3) Star and C41 (DE3) at three different temperatures / incubation times (3-4 hr at 37 °C, 5 hr at 30 °C and overnight at 20 °C). Before cloning, check for the existence of an internal thrombin cleavage site by using the ExPASy PeptideCutter tool31 to prevent cleavage of the protein of interest. Alternatively, there are vectors available providing a cleavage site for HRV3C (i.e. EMBL's pETM-1432) or TEV protease (i.e. EMBL's pETM-11).
Protein purification
For rPPEP-1 constructs cell lysis is performed via sonication on ice/water. For more sensitive proteins that tend to aggregate or precipitate a more "gentle" cell lysis method involving a cell disruptor might be used. Check for large amounts of insoluble protein after the first centrifugation step, which are not detected when using lysis method such as chemical cell lysis. For the purification of rPPEP-1 usually 2 ml of the NiNTA resin were used. Read the manufacturer's instructions on how large the protein binding capacity of the chosen resin is and adjust accordingly. In the first purifications of rPPEP-1, where only 1 ml of resin was used, a lot of protein was not bound to the column and was found in the flow-through (Figure 1D). On the other hand using too much resin might lower the purity of the purified protein. Adjust the imidazole concentration to the binding affinity of the used His-tagged construct to the NiNTA matrix. A stepwise washing procedure is preferred here (i.e. 10 mM, 30 mM, 50 mM, 70 mM imidazole), in which the A280 is monitored and is allowed to reach the baseline during each step. In that way no protein is lost, even if it would elute during a high imidazole wash step (e.g. at 50 or 70 mM). In the case of NHis-rPPEP-1 some protein already elutes at 30 mM imidazole, but it is unclear what kind of protein species it represents. Some proteins precipitate/aggregate when imidazole is dialyzed or diluted out of the protein solution. In such cases reduce the concentration of imidazole in the elution buffer to 150 mM and the time of exposure to high imidazole concentrations, e.g. decrease the concentration of imidazole by elution into a beaker containing a 5-10-fold volume of buffer without imidazole compared to the planned elution volume. For NHis-rPPEP-1 incubation with 2 units thrombin per mg of protein overnight at 4 °C are sufficient to cleave the His-tag to almost 100%. Adjust the amount of thrombin for the protein of interest by using 1-10 units per mg protein at 4 °C to 20 °C. When concentrating the protein using an centrifugal ultrafiltration unit pause in intervals of 5-10 min and mix the protein to homogenize the concentration gradient building up in the concentrator. Otherwise highly concentrated protein could aggregate/precipitate.
Crystallization
rPPEP-1 constantly produces highly intergrown crystals (single crystals growing on top of each other, thus having multiple crystal lattices) that are not suitable for X-ray diffraction analysis. The explanation might be that too many nucleation events are happening in the nucleation zone of the phase diagram (Figure 7).
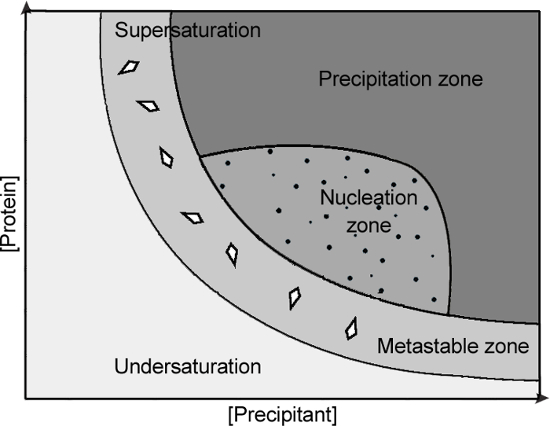
Figure 7: Phase diagram of a protein crystallization experiment. Crystals can only form, when the protein is supersaturated. Nucleation takes place in the nucleation zone and crystal growth in the metastable zone. When a protein is undersaturated, the drop will remain clear.
Lowering of protein or precipitant concentrations does not improve the situation as then no nucleation of rPPEP-1 is observed anymore and the drops stay clear. Microseeding was the method of choice, as tiny nuclei are brought directly into the metastable zone, in which rPPEP-1 crystals eventually grow (Figure 7). By designing the optimization screen in a way that the original condition (2.4 M ammonium phosphate dibasic, 0.1 M Tris pH 8.5) is located at the position C5 (Figure 4) (rather the high end of pH and precipitant concentration) most of the new conditions correspond to a condition with lower supersaturation with a high propensity to represent a part of the metastable zone16 (Figure 7). Thus, the nuclei that are brought in during seeding can potentially grow to large single crystals. To optimize that procedure (amount and size of newly formed crystals) the original seed stock can be diluted 10-2 to 10-5. Here several dilutions of the seed stocks could be tested to determine the best seed dilution for production of large single crystals. Alternatively streak seeding using an animal whisker or tail hair (rabbit, cat, chinchilla or horse) could be employed33.
The procedure can be used to co-crystallize product-peptides and substrate-peptides of rPPEP-1. Crystals in space group P21 diffracting up to 1.25 Å were obtained from seeds stocks diluted 1:250 in the optimization procedure. Crystals grow in the two space groups P212121 (unbound protein) and P21 (complex structures), while originating from very similar conditions within the ammonium phosphate screen of the optimization procedure. Due to the crystal packing found in both r-PPEP-1 crystal forms, principally all small molecules and peptides addressing the unprimed side of the active site alone could be soaked into the crystals. This side of the molecule is accessible from the solvent present in the crystal. However molecules addressing both sub-sites or the primed side only need to be co-crystallized with rPPEP-1, as S-loop opening would be required to accommodate them in the active site. Additionally, rPPEP-1 is contacted by the neighboring molecules at the exit from the substrate-binding site near the S3'site (promotion of crystal contacts in this area) — a fact that also limits the length of substrate peptides to 3 residues at the primed side in these two crystal forms.
Crystal mounting and cryo-protection
Mounting of protein crystals is a method that requires some skill in manipulation under the stereomicroscope and thus needs some practice. Choose the optimal length of nylon loop (or other loop of choice, e.g. litholoop) to prevent the excess of solvent around the crystal that contributes to background scattering and thus a lower signal to noise ratio of the diffraction data. The optimal loop size/length also makes fishing of the crystals easier as the crystal does not slip through the loop. For rPPEP-1 crystals, which are about 100-200 µm in the longest dimension, nylon loops of 0.1-0.2 mm size were chosen. The cryo-protectant may also contribute to worsening of data quality, as the crystal may encounter an osmotic shock when transferred to the cryo-condition. This may hamper or even destroy the inner order of the crystal. Carefully select the type and concentration of the cryo-protectant. rPPEP-1 crystals were cryo-protected with either 20% glycerol or 30% sucrose. If crystallizing a substrate-peptide complex or a complex of rPPEP-1 with another ligand, sucrose should be chosen as cryo-protectant as glycerol binds to the primed site of the substrate-binding site of rPPEP-114.
Structure determination
The AutoSol solution obtained a BAYES-CC of 49.2 ± 18.4, a FOM (figure of merit) of 0.41, a skew of 0.17 and a correlation RMS of 0.85. The model map correlation is 0.86 and the R/Rfree factors are 0.21/0.24. All these parameters indicate a successful structure solution.
