Manufacturing Chimeric Antigen Receptor (CAR) T Cells for Adoptive Immunotherapy
Summary
We describe an approach to reliably generate chimeric antigen receptor (CAR) T cells and test their differentiation and function in vitro and in vivo.
Abstract
Adoptive immunotherapy holds promise for the treatment of cancer and infectious disease. We describe a simple approach to transduce primary human T cells with chimeric antigen receptor (CAR) and expand their progeny ex vivo. We include assays to measure CAR expression as well as differentiation, proliferative capacity and cytolytic activity. We describe assays to measure effector cytokine production and inflammatory cytokine secretion in CAR T cells. Our approach provides a reliable and comprehensive method to culture CAR T cells for preclinical models of adoptive immunotherapy.
Introduction
Chimeric antigen receptors (CARs) provide a promising approach to redirect T cells against distinct tumor antigens. CARs are synthetic receptors that bind an antigen target. While their precise composition is variable, CARs generally contain 3 distinct domains. The extracellular domain directs binding to a target antigen and is typically comprised of a single chain antibody fragment linked to the CAR via an extracellular hinge. The second domain, commonly derived from the CD3ζ chain of the T cell receptor (TCR) complex, promotes T cell activation following CAR engagement. A third costimulatory domain is included to enhance T cell function, engraftment, metabolism, and persistence. The success of CAR T cell therapy in various hematopoietic malignancies including B cell acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL) and multiple myeloma highlights the therapeutic promise of this approach1,2,3,4,5,6. The recent Food and Drug Administration (FDA) approvals for two CD19-specific CAR T cell therapies, tisagenlecleucel for pediatric and young adult ALL and axicabtagene ciloleucel for diffuse large B-cell lymphoma, reinforces the translational merit of CAR T cell Therapy.
CAR T-based approaches involve the isolation of T cells from peripheral blood, activation, genetic modification, and expansion ex vivo. Differentiation is an important parameter regulating CAR T cell efficacy. Accordingly, restricting T cell differentiation during ex vivo culture enhances the ability of the infused product to engraft, expand, and persist, providing long term immunosurveillance following adoptive transfer2,7,8,9. T cells consist of several distinct subsets including: naïve T cells (Tn), central memory (Tcm), effector memory (Tem), effector differentiated (Tte) and stem cell memory (Tscm). Effector differentiated T cells have potent cytolytic ability; however, they are short lived and engraft poorly10,11,12. In contrast, T cells with a less-differentiated phenotype including naïve T cells and Tcm exhibit superior engraftment and proliferative abilities following adoptive cell transfer13,14,15,16,17,18. The composition of the collected T cells in the premanufactured product can vary across patients and correlates with the therapeutic potential of CAR T cells. The proportion of T cells with a naïve-like immunophenotype in the starting apheresis product is highly correlated with both engraftment and clinical response19.
Culture duration is an important parameter influencing differentiation in CAR T cells prepared for adoptive transfer. We recently developed an approach to generate superior quality CAR T cells using an abbreviated culture paradigm20. Using our approach, we showed that limited culture gives rise to CAR T cells with superior effector function and persistence following adoptive transfer in xenograft models of leukemia. Here, we present the approaches to reliably generate CART19 cells (autologous T cells engineered to express anti-CD19 scFv attached to CD3ζ and the 4-1BB signaling domains) and include a detailed description of the assays that provide insight into CAR T bioactivity and efficacy prior to adoptive transfer.
Protocol
All animal studies are approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
1. T Cell Activation, Transduction, and Expansion
- Activate fresh or cryopreserved primary human T cells by mixing with anti CD3/CD28 magnetic beads (e.g., dynabeads) at a ratio of 3 beads per T cell in 6-well cell culture dishes. Culture T cells in X-VIVO 15 medium supplemented with 5% normal human AB serum, 2 mM L-glutamine, 20 mM HEPES, and IL2 (100 units/mL). Maintain T cells at a concentration of 106 T cells/mL during expansion. Culture T cells at 37 °C, 20% O2, and 95% humidity with 5% CO2.
- After overnight stimulation, add lentiviral supernatant to activated T cells. Calculate the volume of supernatant necessary to achieve a multiplicity of infection (MOI) of 3−5.
NOTE: The CD19-BBζ CAR lentivirus plasmid consists of a CD8 hinge, 4-1BB costimulatory domain, and CD3ζ signaling domain21. CD19-BBζ lentiviral supernatant was generated as previously described21. - On day 3, collect a representative aliquot of cells for cryopreservation. Prior to cryopreservation, remove the magnetic beads by gentle pipetting and magnetic separation. Prepare freezing medium containing phosphate-buffered saline (PBS) with 0.5% dimethyl sulfoxide (DMSO) and store in 4 °C until use.
- Centrifuge T cells at 300 x g for 5 min. Discard the supernatant and add 5 mL of PBS. Centrifuge cells at 300 x g for 5 min and discard the PBS.
- Resuspend the cell pellet in 1 mL of cold cryopreservation medium. Freeze T cells in a chilled freezing container and store at -80 °C for 48 h. Transfer the frozen cells to liquid nitrogen.
- Wash the rest of the T cells once in 5 mL of PBS to eliminate residual vector. Centrifuge at 300 x g for 5 min. Decant the PBS and resuspend the cell pellet in T cell culture medium at a concentration of 0.5 x 106 cells/mL.
- Split the T cells into two cultures, designated for day 5 and day 9. Count T cells by flow cytometry using counting beads (Table of Materials) and monoclonal antibodies to human CD4 and CD8, as well as a viability dye (Table of Materials).
- To measure T cell concentration, prepare a master mix containing 500 µL of PBS, 5 µL of counting beads, 10 µL of 7-Amino-actinomycin D cell viability solution, 4 µL of CD4-FITC and 4 µL of CD8-APC. Add 40 µL of T cells to the master mix and measure cell concentration by flow cytometry based on number of live T cells/bead counts. Refeed to maintain the cultures at a concentration of 0.5 x 106 cells/mL every other day.
- On day 5, count and cryopreserve day 5 cultures as described in steps 1.3 and 1.5.
- On day 7, wash 0.5 x 106 T cells in PBS and resuspend in 100 µL of fluorescence activated cell sorting (FACS) buffer. Detect CAR surface protein expression by immunostaining with a fluorescently-conjugated anti-CAR19 idiotype by flow cytometry.
- On day 9, count day 9 cultures and cryopreserve as described in step 1.3.
2. Phenotypic Assessment of T Cell Differentiation
- Prepare a master mix containing pre-titrated antibodies for anti-CD3–BV605 (clone OKT3), anti-CD14–Pacific Blue (PB) (clone HCD14), anti-CD19–PB (clone HIB19), anti-CD4–BV510 (clone OKT4), anti-CD8–H7APC (clone SK1), anti-CCR7–FITC (clone 150503), anti-CD45RO–PE (clone UCHL1), anti-CD27–PE-Cy7 (clone 1A4CD27), anti-CD95–PerCP-Cy5.5 (Clone DX2), and anti-CAR19-APC.
- Prepare individual fluorescence minus one (FMO) controls for anti-CD45RO–PE, anti-CCR7–FITC, anti-CD27–PE-Cy7 and anti-CD95–PerCP-Cy5.5 to distinguish positively stained cells from background.
- Prepare dead cell staining solution by diluting live/dead stock reagent (Table of Materials) 1:10,000 in PBS.
- Thaw day 3, day 5, and day 9 T cells that were previously cryopreserved. Centrifuge 1 x 106 T cells from each group at 300 x g for 3 min. Discard the supernatant. Wash the cells once with PBS. Centrifuge at 300 x g for 3 min and discard the PBS.
- Mix T cells with dead staining solution for 15 min at room temperature (RT), protected from the light.
- Add 1 mL of FACS buffer to quench the dead cell staining dye. Centrifuge at 300 x g for 3 min, discard the supernatant and resuspend the cell pellet in 100 µL of FACS buffer containing the antibody cocktail described in step 2.1 and 2.2. Incubate for 1 h at 4 °C.
- Add 1 mL of FACS buffer and centrifuge at 300 x g for 3 min to wash off unbound antibody. Repeat three times with FACS buffer.
- Resuspend cells in 1% paraformaldehyde (PFA) and store at 4 °C.
- As CD45RO expression decreases after fixation, analyze the samples by flow cytometry after immunostaining. To assess differentiation, gate in the following order: singlets (FSC-H vs FSC-A), live CD3+ T cells (Dump [live-dead violet, CD14-PB and CD19-PB vs CD3-BV605], CD4-BV510 vs CD8-APC-H7). In CD4+ and CD8+ subsets, gate on CD45RO-PE vs CCR7-FITC to define naïve-like T cells (CD45RO–CCR7+), Tcm (CD45RO+CCR7+), Tem (CD45RO+CCR7–), and Tte (CD45RO–CCR7–). To identify Tscm, gate on CD27+ T cells in naïve-like T cells population. In this compartment, Tscm are CD95+ and Tn are CD95–.
3. In Vitro Functional Analysis
- CAR T cell proliferation and cytokine secretion
- Verify CAR expression as well as cell viability as described in steps 1.5 and 1.7, by flow cytometry.
- Wash 5 x 106 T cells from each group (day 3, day 5 and day 9) with PBS and resuspend in a 1 µM solution of carboxyfluorescein diacetate succinimidyl ester (CFSE) in PBS for 3.5 min at RT.
- Immediately add 10 mL of PBS containing 10% fetal bovine serum (FBS) to quench the reaction.
- Centrifuge the solution at 300 x g for 3 min. Discard the supernatant and repeat this wash step three times. Count the cells at the conclusion of CFSE staining using a Coulter counter (Table of Materials).
- Harvest CFSE-stained cells (unstimulated), resuspend in 1% PFA and store at 4 °C for analysis by flow cytometry.
- Incubate the desired number of CFSE-stained CAR T cells with irradiated K562-CD19 (target) as well as K562-wild type (control) cells at a ratio of 1:1 for 120 h in cytokine free culture medium and culture conditions as described in step 1.1.
- After 24 h, centrifuge the culture vessel at 300 x g for 5 min. Collect 120 µL of cellular supernatant. Assess activation-dependent production of IL2, IFNγ, TNFα, GM-CSF, and other inflammatory cytokines (IL1β, IL4, IL5, IL6, IL8, and IL10) by Luminex analyses in accordance with the manufacturer's recommendations.
- Replace the volume of supernatant that was collected in step 3.1.7 with an equivalent volume (120 µL) of fresh medium.
- On day 3 (i.e., after 96 h), count and re-feed at a concentration of 0.5 x 106 cells/mL using bead-based flow cytometry as previously in step 1.5.
- On day 5, count the live CD3+ cells and calculate the fold change of live T cells relative to the live T cell count at day 0.
- Perform a comprehensive analysis of CFSE dilution in dividing CAR T cells using FlowJo software to highlight successive rounds of cell division.
NOTE: Proliferation assays are well described in the FlowJo manual.
- Cytotoxicity assay
NOTE: The ability of CART19 cells to kill target cells expressing CD19 is evaluated using a 51Cr release-assay.- Label the target cells by mixing 5 x 105 K562-CD19, K562-wild type control cells, or NALM6 leukemia cells with 50 µL of Na251CrO4 and 0.5 mL of RPMI supplemented with 10% FBS for 90 min in the incubator at 37 °C.
- Centrifuge cells at 300 x g for 2.5 min. Discard the radioactive supernatant in appropriate disposal bins and wash the target cells in 5 mL of PBS. Repeat the wash steps twice.
- Resuspend the target cells in phenol red-free medium containing 5% FBS. Use this medium for the rest of the procedure to reduce background.
- After evaluating CAR expression and cell viability by flow cytometry (as described in section 5.1), mix CAR T cells with labeled target cells at effector:target (E:T) ratios of 10:1, 3:1 and 1:1, in triplicate. Transfer to a 96-well U bottom plate.
- In parallel, include target cells alone, and target cells with 1% sodium dodecyl sulfate (SDS), to determine spontaneous (S) and maximum (M) 51Cr release, respectively.
- Centrifuge cells at 300 x g for 5 min and incubate for 4 h or 20 h in a 37 °C incubator with 5% CO2.
- After the designated time, centrifuge the culture vessel at 300 x g for 5 min. Collect 35 µL of cellular supernatant and transfer to a reader plate. Avoid bubbles. Let the plate dry overnight.
- Seal the plate with a standard plate seal and count with a liquid scintillation counter. Chromium abundance in the supernatant provides a proxy of target cell killing. Calculate the percentage of specific lysis as follows: 100 x (counts per minute [cpm] experimental release – cpm S release)/ (cpm M release – cpm S release).
4. In Vivo Functional Analysis
- Obtain 6−10-week-old NOD-SCID γc–/– (NSG) mice, which lack an adaptive immune system, and assign them to treatment/control group randomly.
- Inject animals intravenously via tail vein with 1 x 106 NALM6 cells in 0.1 mL sterile PBS.
- After 5−7 days, confirm tumor engraftment by bioluminescence imaging (BLI). Inject 150 mg/kg of D-luciferin to mice which have been anesthetized with isoflurane (volume is dependent on body mass).
- Measure bioluminescence values using an imaging system. Quantify total flux using the corresponding software by drawing rectangles of identical area around mice, reaching from head to 50% of the tail length. Subtract the background for each image individually.
- After establishing leukemia, inject 3 x 106 day 9 CART19 cells, 0.5 x 106 day 3 CAR T cells, or corresponding non-transduced (NTD) human T cells via tail vein in a volume of 100 µL of sterile PBS/Ca2+.
NOTE: As the bioactivity of cells harvested at various intervals during the culture process is different, the chosen concentration of day 3 cells is lower than day 9. - To determine disease progression, measure the bioluminescence values twice a week as described above in step 4.3.
- To determine CAR T cell engraftment, collect 75 µL blood via retro-orbital bleeding in an EDTA-coated tube. Transfer 50 µL of blood to absolute counting tubes.
- Stain blood with antibodies against CD45, CD4, CD8 and CAR for 30 min at RT. Add 400 µL of 1x FACS lysing solution to the tubes and vortex thoroughly. After staining, analyze surface marker expression by flow cytometry.
NOTE: Other surface markers can be used to assess the differentiation and exhaustion status of the T cells in blood. - To measure cytokines levels in blood, centrifuge blood at 1,200 x g for 30 min at 4 °C to separate serum from the upper layer of blood.
- Collect serum and measure the cytokines with a designated reader plate according to the manufacturer's instructions.
Representative Results
Using the methods described above, we stimulated and expanded T cells for either 3 or 9 days (Figure 1A,B). We also analyzed their differentiation profile, as indicated by the gating strategy outlined in Figure 1C, by measuring the abundance of distinct glycoproteins expressed on the cell surface. We show a progressive shift towards effector differentiation over time during ex vivo culture (Figure 1D). We assessed the effector function and proliferative capacity of CAR T cell in response to antigen. We show that cells that were expanded less (harvested earlier) were functionally superior compared to the cells extensively cultured over a longer duration. Day 3 CART19 cells have enhanced proliferative and cytolytic ability upon re-stimulation with their cognate ligand relative to day 9 (Figure 2A,B).
In a human xenograft moue model of ALL, we compared the potency of CAR T cells harvested at 3 days versus 9 days (Figure 3A). We showed a dose dependent anti leukemic response for the CART19 cells generated for 9 days with a complete response for high dose of 3 x 106 and a loss of efficacy for the low dose of 0.5 x 106. The day 3 CART19 cells showed persistent tumor control in both high and low doses of CART19 cells (Figure 3B). This response was associated with the absolute count of CART19 in the peripheral blood of mice (Figure 3C) which was analyzed based on the protocol described above. These results obtained from our comprehensive assessment of CAR T function provide evidence that that CAR T cells harvested earlier (day 3) outperform CAR T cells harvested on day 9.
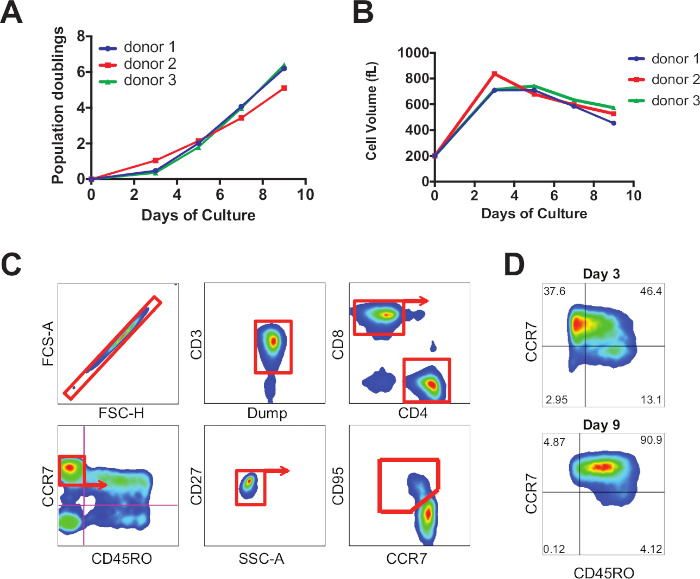
Figure 1: Representative proliferation and differentiation profile of CAR T cells. (A) CART19 cell expansion following stimulation with anti-CD3/CD28 magnetic beads. (B) Cell size was assessed by Coulter analysis throughout the culture. (C) Representative gating strategy for phenotypic analysis of T cells. (D) Temporal analysis of T cell differentiation. Please click here to view a larger version of this figure.
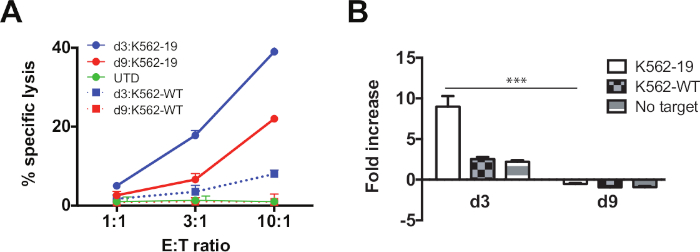
Figure 2: Day 3 CART19 cells display enhanced effector function and proliferation relative to day 9 cells. (A) CART19 cells were harvested on day 3 and 9 and co-cultured at the indicated E:T ratio with CD19-expressing K562 cells (K562-19) or wild-type K562 (K562-wild type). Specific cytotoxicity was measured by 51Cr release after 4 h. (B) CSFE-labeled CART19 cells were co-cultured with K562-19, K562-wild-type, or medium only for 120 h at a 1:1 E:T ratio. Cells were harvested at indicated timepoints. Absolute counts were assessed by flow cytometry. Relative fold changes of live T cell count normalized to T cell count at day 0 are shown. Data are plotted as mean ± standard deviation (SD). ***P < 0.001 comparing day 3 versus day 9. Please click here to view a larger version of this figure.
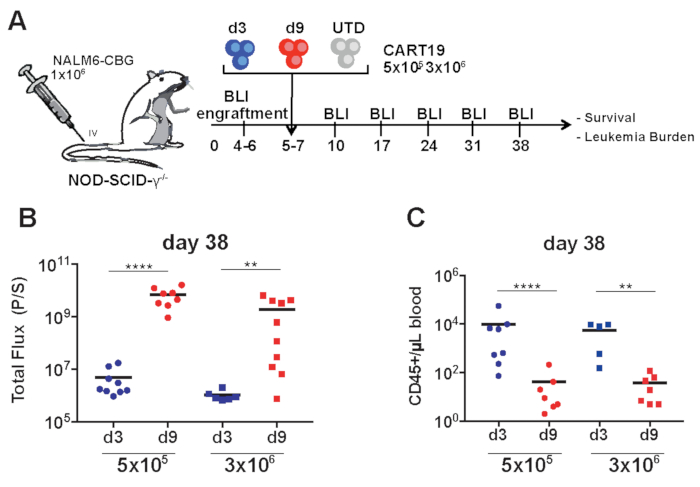
Figure 3: Day 3 CART19 cells are more potent in vivo than day 9 cells. (A) Schematic of the xenograft model and CART19 cell treatment. Day 3 and 9 CART19 cells or control T cells (UTD) were IV-injected in mice 5−7 days after NALM6 injection. (B) Quantification of tumor burden by bioluminescence imaging on day 38 in mice treated with CART19 cells harvested on day 3 and day 9. Symbols represent one mouse each. Horizontal black line: mean of each group. (C) Absolute peripheral blood CD45+ T cell counts were measured every two weeks after CART19 cell injection and at the end of the experiment by an appropriate counting method (such as TruCount) Unpaired Mann-Whitney test, two-tailed was used. **P < 0.01, ***P < 0.001, ****P < 0.0001. Please click here to view a larger version of this figure.
Discussion
Here we describe approaches to measure the function and efficacy of CAR T cells harvested at varying intervals throughout ex vivo culture. Our methods provide comprehensive insight into assays designed to assess proliferative capacity as well as effector function in vitro. We describe how to measure CAR T cell activity following stimulation through the CAR and detail xenograft models of leukemia using CAR T cells harvested at day 3 vs day 9 of their logarithmic expansion phase.
There are inherent challenges in comparing the efficacy of CAR T cells harvested at different time-points during ex vivo expansion. As the quantity of T cells generated over a 3-day culture duration is low, there may be insufficient numbers to perform a comprehensive assessment of function. This is exacerbated in the context of patient T cells whose proliferative ability is often diminished due to extrinsic and intrinsic factors20.
How many CAR T cells should be infused given that day 3 cells exhibit enhanced metabolic and proliferative ability compared to their day 9 counterparts that are exiting their logarithmic proliferative phase? We estimated what numbers of day 3 CAR T cells would reach 3 x 106 if they were expanded for a further 6 days in culture. We used this estimate to inform how many day 3 CAR T cells should be infused (0.5 x 106) to compare equivalently to day 9. Accordingly, we apply the “stress test” approach to compare the bioactivity, efficacy, and persistence of infused CAR T cells. Decreasing the number of infused CAR T cells from 3 x 106 to 0.5 x 106 reveals differences that would otherwise be masked by saturation of numbers. Mechanistically, tumor control relies on the accumulation of sufficient effector cells to lyse their corresponding target cells. At high numbers of infusion, both day 3 and day 9 CAR T cells exhibit functional competence.
Another challenge in working with day 3 CAR T cells is their firm attachment to the stimulatory surface. Displacing them from the magnetic beads requires repetitive pipetting to mechanically dissociate them and enhance their recovery from culture. In contrast, day 9 cells have 1) already detached from the beads, and 2) diluted the beads to such an extent that they can be harvested with relative ease.
Another important variable in the CAR T cell manufacturing process is the choice of cell culture medium. RPMI-based medium which has been supplemented with FBS is commonly used for experimental purposes. In contrast, either X-VIVO 15 or OpTmizer, supplemented with human serum are preferred in clinical applications. While these are less characterized, they may contain components that facilitate T cell expansion in a shorter time period. Their impact on differentiation is unknown. Additionally, the addition of cytokines influences growth, survival, and phenotype. While IL-2 drives rapid proliferation and differentiation into effector cells, IL-7 and IL-15, which originate in the lymph node and have known roles in homeostatic persistence, improves expansion of T cells and promote a memory stem/central memory phenotype22,23,24,25.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported in part through funding provided by Novartis Pharmaceuticals through a research alliance with the University of Pennsylvania (Michael C. Milone) as well as St. Baldrick's Foundation Scholar Award (Saba Ghassemi).
Materials
| Anti CD3/CD28 dynabeads | Thermo Fisher | 40203D | |
| APC Mouse Anti-Human CD8 | BD Biosciences | 555369 | RRID:AB_398595 |
| APC-H7 Mouse anti-Human CD8 Antibody | BD Biosciences | 560179 | RRID:AB_1645481 |
| BD FACS Lysing Solution 10X Concentrate | BD Biosciences | 349202 | |
| BD Trucount Absolute Counting Tubes | BD Biosciences | 340334 | |
| Brilliant Violet 510 anti-human CD4 Antibody | BioLegend | 317444 | RRID:AB_2561866 |
| Brilliant Violet 605 anti-human CD3 Antibody | BioLegend | 317322 | RRID:AB_2561911 |
| CellTrace CFSE Cell Proliferation Kit | Life Technolohgies | C34554 | |
| CountBright Absolute Counting Beads, | Invitrogen | C36950 | |
| FITC anti-Human CD197 (CCR7) Antibody | BD Pharmingen | 561271 | RRID:AB_10561679 |
| FITC Mouse Anti-Human CD4 | BD Biosciences | 555346 | RRID:AB_395751 |
| HEPES | Gibco | 15630-080 | |
| Human AB serum | Valley Biomedical | HP1022 | |
| Human IL-2 IS, premium grade | Miltenyi | 130-097-744 | |
| L-glutamine | Gibco | 28030-081 | |
| Liquid scintillation counter, MicroBeta trilux | Perkin Elmer | ||
| LIVE/DEAD Fixable Violet | Molecular Probes | L34964 | |
| Multisizer Coulter Counter | Beckman Coulter | ||
| Na251CrO4 | Perkin Elmer | NEZ030S001MC | |
| Pacific Blue anti-human CD14 Antibody | BioLegend | 325616 | RRID:AB_830689 |
| Pacific Blue anti-human CD19 Antibody | BioLegend | 302223 | |
| PE anti-human CD45RO Antibody | BD Biosciences | 555493 | RRID:AB_395884 |
| PE/Cy5 anti-human CD95 (Fas) Antibody | BioLegend | 305610 | RRID:AB_493652 |
| PE/Cy7 anti-human CD27 Antibody | Beckman Coulter | A54823 | |
| Phenol red-free medium | Gibco | 10373-017 | |
| UltraPure SDS Solution, 10% | Invitrogen | 15553027 | |
| Via-Probe | BD Biosciences | 555815 | |
| X-VIVO 15 | Gibco | 04-418Q | |
| XenoLight D-Luciferin – K+ Salt | Perkin Elmer | 122799 |
References
- Brentjens, R. J., et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Science Translational Medicine. 5 (177), 177ra138 (2013).
- Grupp, S. A., et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. The New England Journal of Medicine. 368 (16), 1509-1518 (2013).
- Kalos, M., et al. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Science Translational Medicine. 3 (95), 95ra73 (2011).
- Kochenderfer, J. N., Rosenberg, S. A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nature Reviews. Clinical Oncology. 10 (5), 267-276 (2013).
- Maude, S. L., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England Journal of Medicine. 371 (16), 1507-1517 (2014).
- Porter, D. L., Levine, B. L., Kalos, M., Bagg, A., June, C. H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England Journal of Medicine. 365 (8), 725-733 (2011).
- Porter, D. L., et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science Translational Medicine. 7 (303), 303ra139 (2015).
- Maude, S. L., Teachey, D. T., Porter, D. L., Grupp, S. A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 125 (26), 4017-4023 (2015).
- Kochenderfer, J. N., et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. Journal of Clinical Oncology. 35 (16), 1803-1813 (2017).
- Bollard, C. M., Rooney, C. M., Heslop, H. E. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nature Reviews. Clinical Oncology. 9 (9), 510-519 (2012).
- Brestrich, G., et al. Adoptive T-cell therapy of a lung transplanted patient with severe CMV disease and resistance to antiviral therapy. American Journal of Transplantation. 9 (7), 1679-1684 (2009).
- Savoldo, B., et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs). Blood. 108 (9), 2942-2949 (2006).
- Berger, C., et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. The Journal of Clinical Investigation. 118 (1), 294-305 (2008).
- Gattinoni, L., et al. A human memory T cell subset with stem cell-like properties. Nature Methods. 17 (10), 1290-1297 (2011).
- Hinrichs, C. S., et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proceedings of the National Academy of Sciences of the United States of America. 106 (41), 17469-17474 (2009).
- Klebanoff, C. A., et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proceedings of the National Academy of Sciences of the United States of America. 102 (27), 9571-9576 (2005).
- Wang, X., et al. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 117 (6), 1888-1898 (2011).
- Wang, X., et al. Comparison of naive and central memory derived CD8+ effector cell engraftment fitness and function following adoptive transfer. Oncoimmunology. 5 (1), e1072671 (2016).
- Fraietta, J., et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nature Medicine. 24 (5), 563-571 (2018).
- Ghassemi, S., et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunology Research. 6 (9), 1100-1109 (2018).
- Milone, M. C., et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Molecular therapy : the journal of the American Society of Gene Therapy. 17 (8), 1453-1464 (2009).
- Cieri, N., et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 121 (4), 573-584 (2013).
- Cui, G., et al. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8 T Cell Longevity. Cell. 161 (4), 750-761 (2015).
- Xu, Y., et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 123 (24), 3750-3759 (2014).
- Singh, N., Perazzelli, J., Grupp, S. A., Barrett, D. M. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Science Translational Medicine. 8 (320), 320ra323 (2016).
