In the present study, parental populations of C. elegans (P0) were infected at the L1 stage with a low dose of N. parisii spores. These infection conditions are typically used to obtain high numbers of microsporidia-resistant F1 progeny through bleaching of the parents. Infected parental populations and uninfected controls were fixed at 72 hpi and stained with DY96 to visualize the worm embryos and microsporidia spores (Figure 1A). Infected animals are small, contain many microsporidia spores, and produce fewer embryos than healthy uninfected controls. Assessment of worm gravidity showed that ~95% of uninfected animals produced offspring, compared to less than 80% of the infected animals (Figure 1B). Quantifications revealed that ~90% of the microsporidia-treated population were infected, as determined by the number of worms containing DY96-stained spores (Figure 1C). To obtain immune-primed F1 progeny, it is important to use a dose of microsporidia that is high enough to ensure most parents are infected but low enough to ensure that the population can still produce progeny. A table of the microsporidia doses used to obtain the data in this study is provided (Table 1).
Uninfected and infected parent populations (Figure 1) were treated with sodium hypochlorite at 72 hpi to obtain naïve and immune-primed F1 progenies. F1 animals were exposed to a high dose of N. parisii spores at the L1 stage to test for inherited immunity to microsporidia. At 72 hpi, F1 animals were fixed and stained with DY96 to assess microsporidia resistance (Figure 2A). Quantifications of these fixed animals revealed that the primed worms contained significantly more embryos than their naïve counterparts, indicating greater fitness in the face of infection (Figure 2B). FIJI/ImageJ was used to determine the parasite burden of individual naïve and immune-primed worms (i.e., percentage of the body filled with fluorescent N. parisii spores) (Figure 2C). Quantifications revealed a dramatic reduction in the parasite burden of worms that came from infected parents (Figure 2D). Further, calculations of individual worm size revealed that primed worms had a significant growth advantage over naïve animals in the face of N. parisii infection (Figure 2E). These data demonstrate that N. parisii-infected parents produce offspring with high levels of microsporidia resistance.
Previous work has revealed that inherited immunity to microsporidia reduces invasion of intestinal cells by N. parisii19. To visualize differences in host cell invasion, naïve and immune-primed animals (obtained from uninfected or infected parents, as above) were exposed to a maximal dose of N. parisii at the L1 stage. At 30 dpi, the worms were fixed and co-stained, using a FISH probe to detect N. parisii RNA and DY96 to detect N. parisii spore walls. Imaging revealed that, while naïve animals typically contained multiple spores and several infected cells (sporoplasms), the primed animals had far fewer (or no) spores and typically no sporoplasms (Figure 3).
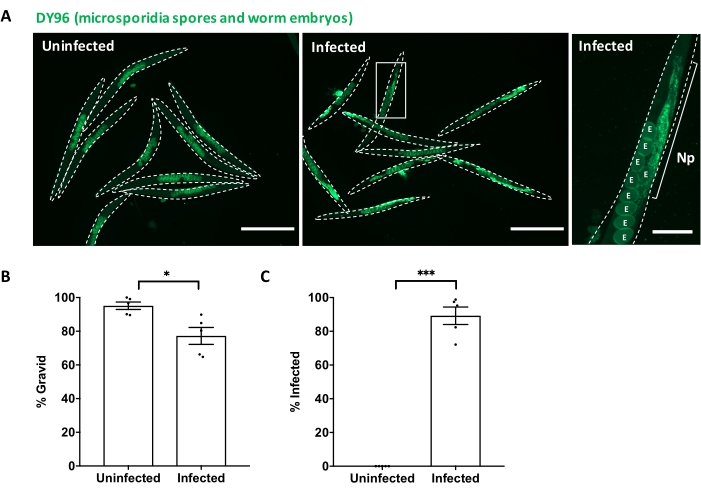
Figure 1: Direct Yellow 96 (DY96) staining of uninfected and N. parisii-infected C. elegans reveals worm gravidity and infection status. (A–C) N2 C. elegans were not infected or infected with a low dose of N. parisii at the L1 stage. At 72 hpi, the worms were fixed, stained with DY96, and imaged to assess worm gravidity and infection status. (A) Representative images are shown. DY96 enables visualization of worm embryos and microsporidia spores (chitin). An inset image of an infected worm is shown on the right-hand side. Embryos are labeled 'E'; N. parisii infection of the intestine is labeled 'Np'. Scale bar for left and middle images = 500 µm. Scale bar for the right image = 100 µm. (B) Worms carrying one or more embryos were scored as gravid and graphed.(C) Worms carrying any number of N. parisii spores in the intestinal cells were scored as infected and graphed. (B–C) Data pooled from 5 independent experiments using n = 100 worms per condition per experiment. Mean ± SEM is shown. The p values were determined by the unpaired two-tailed Student's t-test. Significance was defined as: *p < 0.05; ***p < 0.001. Please click here to view a larger version of this figure.
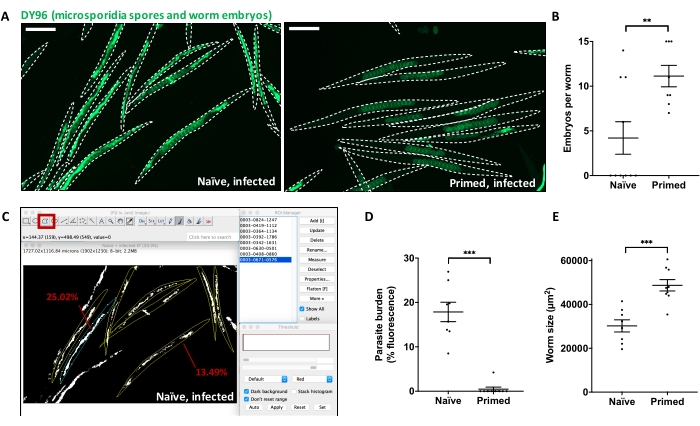
Figure 2: Direct Yellow 96 (DY96) staining of naïve and immune-primed C. elegans to determine embryos per worm, microsporidia burden, and worm size. (A) N2 C. elegans were infected or not infected with a low dose of N. parisii at the L1 stage. At 72 hpi, the worms were treated with sodium hypochlorite to release embryos. The resulting naïve and immune-primed offspring were infected with a high dose of N. parisii at the L1 stage. At 72 hpi, the worms were fixed, stained with DY96, and imaged to visualize worm embryos and parasite burden. Representative images are shown. Scale bar = 200 µm.(B) The number of embryos per worm was counted and graphed from (A). (C) A screenshot from FIJI/ImageJ shows naïve worms from panel A that have been thresholded to visualize parasite burden (shown in white) using the Thresholding window in the bottom right. Here, individual worms were outlined using the "Polygon selections" tool highlighted in red and defined as "Region of interests" (ROI) using the ROI window in the top right. The Analyze > Measure > Area function was used to quantify the parasite burden of two example worms, shown to be 25.02% and 13.49%. (D) The parasite burden per worm was determined and graphed from (A). (E) From the above images, individual worm size was calculated from animals outlined as in panel C using the Analyze > Measure > Area function. (B, D, and E) Mean ± SEM is shown. The p values were determined by unpaired two-tailed Student's t-test. Significance was defined as: *p < 0.05. **p < 0.01, ***p < 0.001. Please click here to view a larger version of this figure.
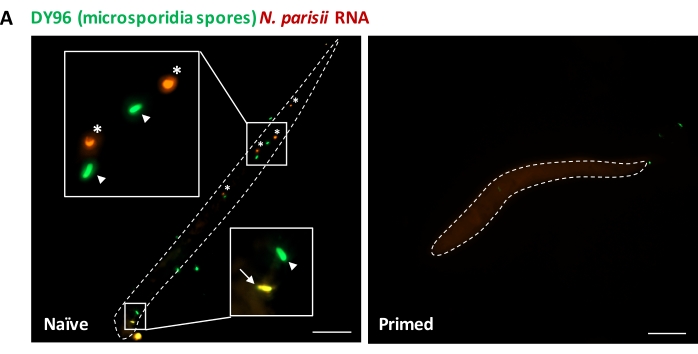
Figure 3: Fluorescence in situ hybridization (FISH) against N. parisii 18S rRNA reveals parasite invasion of C. elegans intestinal cells in naïve but not primed animals. N2 C. elegans were infected or not infected with a low dose of N. parisii at the L1 stage. At 72 hpi, the worms were treated with sodium hypochlorite to release embryos. The resulting naïve and immune-primed offspring were infected with a maximal dose of N. parisii at the L1 stage. At 30 mpi, the worms were fixed, subjected to FISH to detect sporoplasms, stained with DY96 to detect microsporidia spore walls, and imaged. Representative images are shown. Inset images for the naïve worm show sporoplasms (red, asterisks), fired spores (green, arrowheads), and an unfired spore (yellow, arrow). The naïve infected worm shown displays 4 sporoplasms. Scale bar = 20µm. Please click here to view a larger version of this figure.
| N. parisii dose | Plate concentration (spores/cm2) | Millions of spores per 6 cm plate | Millions of spores per 10 cm plate |
| Low | 35,400 | – | 2.7 |
| High | 88,400 | 2.5 | – |
| Maximal | 2,12,000 | 6 | – |
Table 1: N. parisii doses employed in the study.
