As the iDISCO+ protocol introduces significant tissue shrinkage, which is easily noticeable by eye (Figure 2B), we added an MRI step to this pipeline prior to tissue clearing to quantify the shrinkage induced by tissue clearing. The workflow starts with removal of the non-brain tissue from the MR image (Figure 2C). Next, a rigid transformation (3 translation and 3 rotation angles) is applied to align the MR image to the light-sheet image (Figure 2D). By doing so, we observed a 60% total volume loss induced by the tissue clearing procedure (Figure 2E), which was consistent with volume change estimates by eye (Figure 2B). This result is quite different from a previous report where a shrinkage of 5%-10%10,32 was reported. The difference in shrinkage may be caused by the difference in animal age between the two studies (P4 vs. adult brains) or due to time differences in the methanol wash in the clearing step of the iDISCO+ protocol (16 h vs. 2 h). To map degrees of tissue shrinkage across different regions of the brain, we further deployed a deformable image registration where the light-sheet image is used as the reference. The registered MR image is shown in (Figure 2F). The estimated volume change due to the tissue clearing procedure is color-coded, where a larger degree of shrinking is observed in the cortex as compared to other brain regions (Figure 2G).
To measure volumes of brain regions, we performed segmentation of the MRI through registration to an annotated atlas. Segmentation of MR-imaged brains was considered successful if the reference atlas overlay closely matched the anatomical boundaries identified by differences in contrast in the raw MRI images (Figure 2H). The overlay during registration is dependent on good sample preparation (Figure 2A), image acquisition, and skull stripping. The skull stripping step is crucial for good registration results as the registration will attempt to warp the reference image from the atlas to the entire MR image, including any skull that remains in the image. Skull inclusion in registration can distort the results and produce an incorrectly registered 3D volume rendering of the segmentation. The final 3D volume rendering can then be visualized using ITK-SNAP33 (Figure 2H). This method is used to assess gross brain volume differences across sample groups. The cellular basis underlying volume differences discovered with MRI can be pursued using cell counting with tissue clearing, lightsheet microscopy, and NuMorph.
The goal of tissue clearing and analysis is to assess the contributions of different cell types to differences across experimental conditions (e.g., genotype, environmental exposure) by counting individual nuclei using NuMorph. Tissue clearing using the iDISCO+ protocol and neuronal layer specific nuclei markers resulted in clearly defined cell groups of upper and lower layer neurons in the isocortex (Figure 3).
Cell counting using NuMorph is dependent on successful preprocessing steps involving intensity adjustment, channel alignment, and stitching (Figure 4A). Errors occurring in any of these steps can result in improper stitching (Figure 4B,C). For instance, improper image acquisition can result in images with in-focus and out-of-focus patterns during preprocessing (Figure 4C). To count nuclei from specific brain regions, the stitched images are annotated using a publicly available atlas. We registered our stitched images to a corrected version of the P4 Allen Developmental Mouse Brain Atlas34. Here, the stitched images were downsampled from a high resolution (0.75 x 0.75 x 4 µm³/voxel) to a lower resolution (25 µm³/voxel isotropic resolution) to match the resolution of the atlas. Matching the resolution of the atlas ensures correct registration of the images to the atlas and provides regional annotations in the acquired images (Figure 5A).
To perform specific cell-type specific counting, we labeled upper layer neurons expressing Brn2, lower layer neurons expressing Ctip2, and all nuclei with TO-PRO-3 (Figure 3B). Imaging is performed at sufficient spatial resolution to separate individual nuclei (0.75 x 0.75 x 4 µm3/voxel). The centroids of nuclei are detected with a trained 3D-Unet model in NuMorph (Figure 5B,C). We detected ~12 × 106 total nuclei that were To-Pro-3+ in the isocortex, including ~2.6 × 106 Brn2+ and 1.6 × 106 Ctip2+ nuclei. We also detected ~3.7 × 106 and 2.9 × 106 To-Pro-3+ total nuclei in the basal ganglia and hippocampal allocortex, respectively. Approximately 1.5 × 106 and <1 × 106 Ctip2+ cells were counted in the basal ganglia and hippocampal allocortex, respectively, although a negligible number of Brn2+ cells were detected (Figure 5D). The total nuclei counts in the isocortex and hippocampal allocortex combined (~15 million cells) are similar to previously reported total cell counts in the cerebral cortex of the mouse15. These results demonstrate the utility of MRI imaging, iDISCO+ tissue clearing methodology, and NuMorph computational analysis to reveal volumetric, total cell count, and cell-type specific counts underlying differences in mouse brain structure across experimental groups.
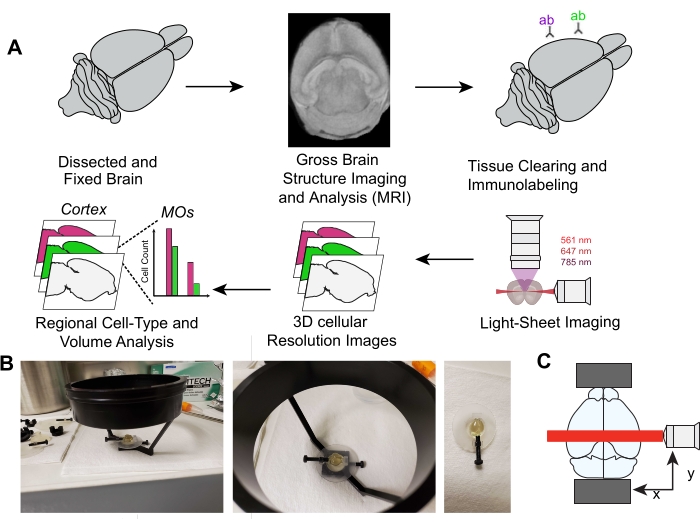
Figure 1: Overview of whole-brain single-cell analysis of intact neonatal 3D tissue cleared mouse brains. (A) Overview of iDISCO+ tissue clearing, light sheet imaging, and 3D image analysis. (B) Images showing correct sample mounting on the light-sheet microscope. (C) Cartoon showing sample mounting relative to light-sheet path. Abbreviation: ab = antibody. Please click here to view a larger version of this figure.
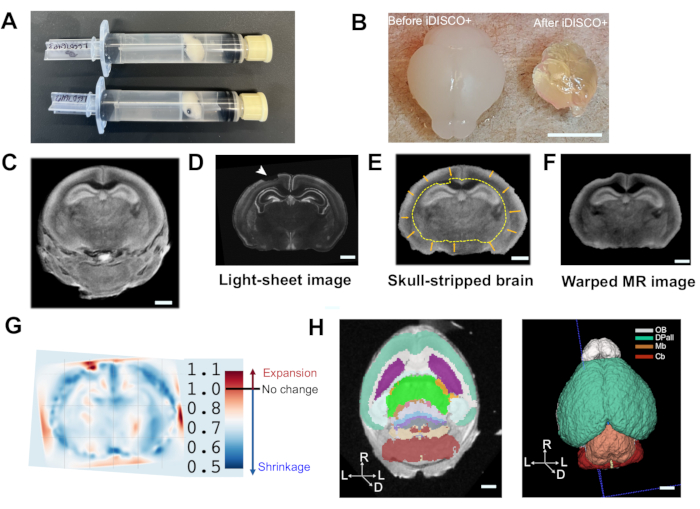
Figure 2: Gross brain structure measurements with MRI. (A) Image showing sample preparation for MRI. (B) Images of representative P4 brains before and after iDISCO+ tissue clearing. Scale bar = 5.0 cm. (C) Raw MR image with skull attached at 60 x 60 x 60 µm³. Scale bar = 800 µm. (D) Light-sheet image of mouse brain at 25 x 25 x 25 µm³. Total volume = 68.7 mm3. Note that a small section of the dorsal isocortex was unintentionally removed during dissection marked by arrowhead (white). Scale bar = 800 µm. (E) MR image of mouse brain after skull stripping and rigid registration. Total volume = 171.9 mm3. Insert (yellow) shows brain contour from the light-sheet image. Scale bar = 800 µm. (F) Registered MR image in reference to light-sheet image to assess deformation. Scale bar = 800 µm. (G) Voxel-wise brain mapping to assess volume changes between light-sheet image and MR image where blue and red indicate shrinkage and expansion from MR image to the light-sheet image, respectively. Overall there was a 60% volume loss, but some regions such as the isocortex showed greater shrinkage relative to others. (H) Left panel: Axial view of MRI image with registered segmentations from the Allen Developmental Brain Atlas overlaid. Right panel: 3D volume rendering of segmentation. Scale bar = 800 µm. Abbreviations: OB = olfactory bulb; Dpall = dorsal pallium/isocortex; Mb = midbrain; Cb = cerebellum. Orientation: R = Rostral; L = Lateral; D = Dorsal. Please click here to view a larger version of this figure.
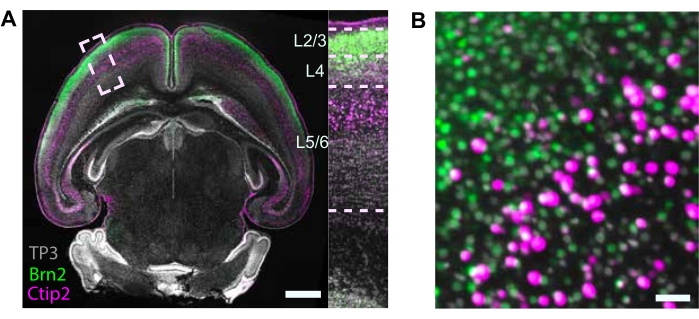
Figure 3: Cellular resolution images and channel alignment. (A) Optical axial section of TO-PRO-3 (TP3) nuclear staining and immunolabeling for Ctip2 (lower layer neuron) and Brn2 (upper layer neuron) markers in P4 mouse brain. Scale bar = 1 mm. (B) Enlarged inset of cortical areas in A (box) showing correct channel alignment with expected localization of Brn2-immunolabeled upper layer and Ctip2-immunolabeled lower layer neurons. Scale bar = 50 µm. Please click here to view a larger version of this figure.
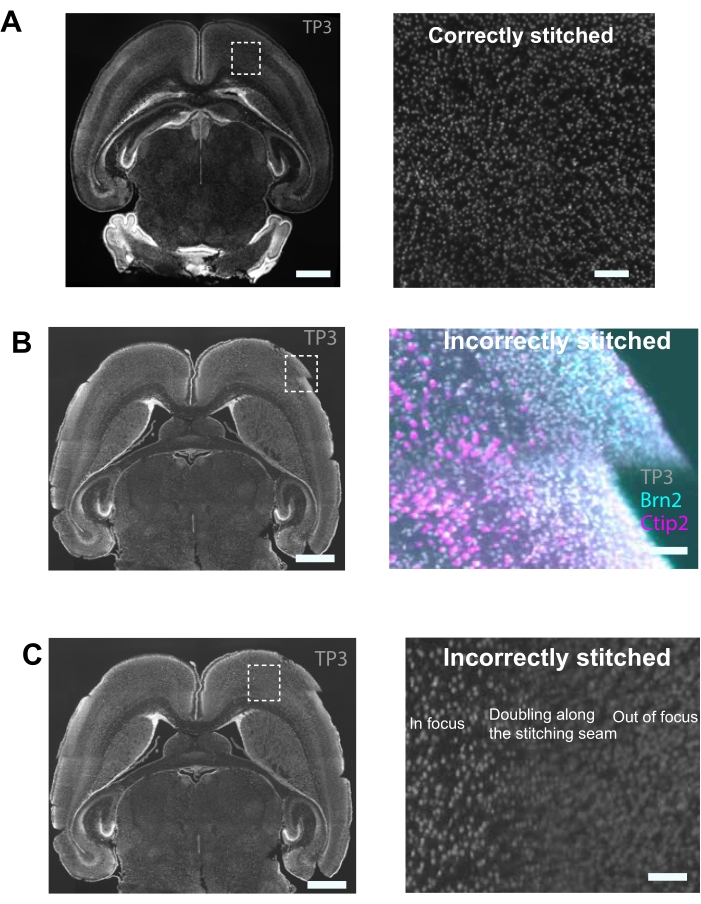
Figure 4: Image stitching examples. (A) Sample results from a 2D iterative, correctly stitched image. Scale bar = 1 mm. Insert shows an example of correct stitching zoomed in. Scale bar = 500 µm. (B) Sample results from a 2D iterative, incorrectly stitched image. Scale bar = 1 mm. Insert shows an example of incorrect stitching zoomed in (overhang). Scale bar = 100 µm. (C) Sample results from a 2D iterative incorrectly stitched image. Scale bar = 1 mm. Insert shows an example of incorrect stitching zoomed in (out-of-focus). Scale bar = 200 µm. Please click here to view a larger version of this figure.
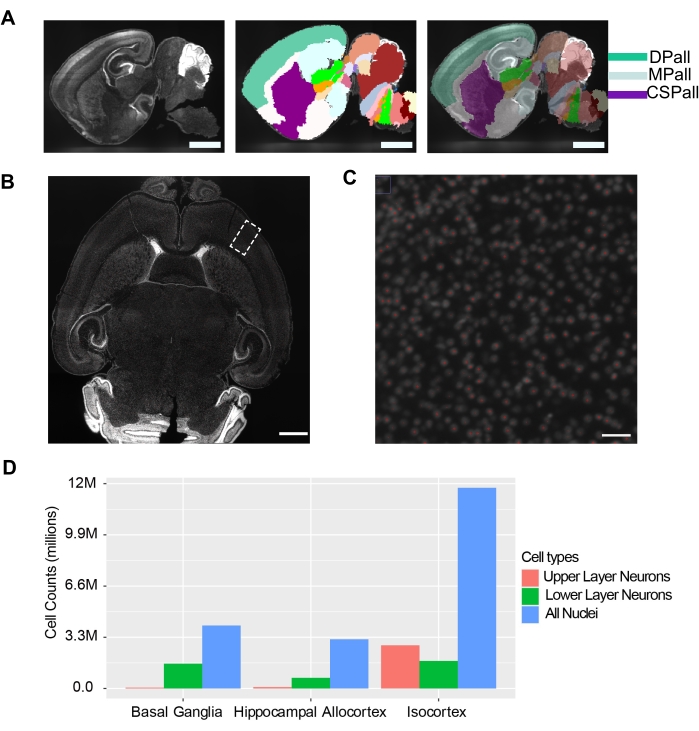
Figure 5: Low resolution image registration, high resolution nuclei detection, and cell counting in the P4 mouse brain. (A) Left panel: An example of a z-slice from whole brain light sheet 3D images resampled to 25 µm isotropic resolution. Scale bar = 1 mm. Middle panel: Annotations from the Allen Developmental Brain Atlas (P4) registered to the microscopy image. Scale bar = 1 mm. Right panel: Overlay of the left and middle panels. Scale bar = 1 mm. (B) Example of an intensity adjusted stitched image in axial view. The boxed region is shown in (C). Scale bar = 1 mm. (C) Automated detection of nuclei (red). Scale bar = 50 µm. (D) Quantification of cell types in different brain regions of the P4 brain (Basal ganglia, Hippocampal Allocortex, and Isocortex). Red = Upper layer neurons labeled with Brn2, Green = Lower Layer neurons labeled with Ctip2, and Blue = all nuclei labeled with To-Pro-3 dye. Abbreviations: DPall = Dorsal pallium (isocortex); MPall = Medial pallium (hippocampal allocortex); CSPall = Central subpallium(classic basal ganglia). Please click here to view a larger version of this figure.
| NuMorph Analysis Stage | Estimated Time |
| Intensity Adjustment | 1 h |
| Channel Alignment | 2 min |
| 2D Iterative Stitching | 12 h |
| Resampling | 3 min |
| Registration | 3 min |
| Cell Counting | 5 h |
| Cell Classification | 10 min |
Table 1: NuMorph analysis times.
