En las últimas dos décadas, la bioterapéutica ha evolucionado hasta convertirse en un pilar de la industria farmacéutica moderna. La pandemia de SARS-CoV2 y otras afecciones potencialmente mortales han aumentado aún más la necesidad de un desarrollo más rápido y amplio de moléculas biofarmacéuticas 1,2,3.
El peso molecular bioterapéutico es crítico para la identificación de la molécula, en combinación con otros ensayos analíticos. Las masas de subunidades intactas y reducidas se utilizan a lo largo de los ciclos de vida de descubrimiento y desarrollo como parte de las estrategias de control destinadas a mantener la calidad, como se describe en el QTPP (Quality Target Product Profile)4.
El desarrollo analítico en la industria biofarmacéutica se basa en gran medida en las mediciones de masa para el análisis de masa intacta y la caracterización profunda mediante el mapeo de péptidos o el monitoreo del método multiatributo (MAM). En el centro de estas técnicas que utilizan plataformas modernas de espectrometría de masas (MS) se encuentra la capacidad de proporcionar mediciones de masa precisas de alta resolución (HR/AM). La mayoría de los instrumentos HR/AM producen precisiones de masa en el rango de 0,5-5 ppm, que se escalan con el rango de masa. La capacidad de medir masas con precisión para moléculas grandes intactas permite la identificación rápida y confiable de terapias de moléculas grandes. Dado que la resolución isotópica no puede alcanzarse utilizando las condiciones experimentales típicas para moléculas grandes (>10 kDa), se deben calcular las masas medias para su comparación e identificación 5,6.
Un espectro típico de masa de proteína intacta o subunidad representa el perfil proteoforme general, que contiene información compuesta sobre las diversas formas moleculares resultantes de las modificaciones postraduccionales (PTM) y cualquier diferencia de estructura primaria, como clips o variantes de secuencia. La naturaleza relativamente fácil y de alto rendimiento de estas mediciones las hace atractivas para la caracterización y como controles de monitoreo durante el proceso 7,8. El análisis de datos para estos experimentos generalmente requiere que el usuario defina el espacio de búsqueda de formas moleculares (rango de PTM u otras formas moleculares). En el caso de las proteínas glicosiladas, este espacio de búsqueda está impulsado en gran medida por la heterogeneidad de los glicoformes. Las combinaciones de múltiples PTM, las configuraciones de enlaces disulfuro y otras variaciones a lo largo de la estructura primaria hacen que el cálculo de todas las formas moleculares posibles sea una tarea tediosa. Por lo tanto, el cálculo manual de las posibles formas moleculares es un proceso que consume tiempo y recursos con un alto potencial de error humano.
Aquí, presentamos una herramienta de cálculo de masa que fue desarrollada considerando las características más importantes de las moléculas bioterapéuticas, tales como mAbs, bsAbs, ADCs, etc. La herramienta permite la fácil incorporación de variables de espacio de búsqueda para el cálculo consistente de masas y composiciones elementales. El carácter modular de esta herramienta permitirá seguir desarrollándola y aplicándola al cálculo de masas y a la adaptación de masas para otras modalidades.
El módulo GUI permite al usuario especificar la entrada para el cálculo de masa, como se muestra en la Figura 1; En concreto, el usuario introduce secuencias de aminoácidos de una sola letra para cadenas de anticuerpos ligeros y pesados. Las modificaciones comunes para la ciclación de N-terminal de cadena pesada y el recorte de lisina C-terminal se incluyen como casillas de verificación. Además, la fórmula química/composición elemental se puede agregar/restar de estas cadenas de proteínas a través del cuadro de texto Chem Mod respectivo. Esto permite al usuario la flexibilidad de agregar una composición elemental que incluye múltiples modificaciones postraduccionales o una carga útil de molécula pequeña en el caso de un ADC. Como la mayoría de los anticuerpos monoclonales terapéuticos están diseñados para eliminar los sitios de glicosilación en la cadena ligera, la glicosilación en la cadena ligera se deja opcional y se puede especificar mediante una casilla de verificación en la interfaz gráfica de usuario.
Una variación típica del análisis de masa intacta para anticuerpos es un análisis de masa de subunidades reducidas, en el que la cadena ligera se separa de la cadena pesada mediante la reducción de los enlaces disulfuro entre cadenas. Dependiendo de la fuerza del agente reductor utilizado, los enlaces disulfuro intracadena pueden o no escindirse. Los usuarios tienen la flexibilidad de introducir el número total de enlaces disulfuro en función del subtipo de IgG o en el caso de unADC 9 conjugado con cisteína.
La aplicación calcula las masas de forma ascendente, en la que las composiciones elementales se calculan primero para las cadenas pesadas y ligeras individuales. A continuación, se tiene en cuenta la ciclación N-terminal de cadena pesada (HC) Lys-clipping ajustando las composiciones elementales calculadas. A continuación, se aplican las modificaciones químicas especificadas a las cadenas pesadas y/o ligeras. Dependiendo del tipo de análisis y de los patrones de enlace disulfuro especificados por el usuario, el número de hidrógenos se ajusta para las dos cadenas polipeptídicas. Las masas de HC glicosilado y de cadena ligera (LC) (opcional) se calculan en función de la entrada del usuario. Finalmente, se combinan múltiples masas HC y LC, y los números de enlace disulfuro se actualizan automáticamente para el cálculo de masa intacta.
Con moléculas más grandes, como proteínas intactas, las masas monoisotópicas no se pueden medir debido al defecto de masa aditivo cuando se utilizan espectrómetros de masas con un poder de resolución típico. En cambio, se miden o informan masas nominales o promedio 5,10,11,12,13. Las masas elementales medias pueden variar en función de la fuente utilizada para las masas seleccionadas14,15. Si bien las diferencias en las masas elementales pueden ser pequeñas, pueden sumar valores significativos para los cálculos de peso molecular de moléculas grandes. Las masas elementales medias utilizadas por defecto en la aplicación de software se muestran en la Tabla Suplementaria 1. Para entornos regulados como el campo de la investigación y el desarrollo (I&D) biofarmacéutico, es importante mantener masas moleculares consistentes porque los cambios en las masas pueden implicar cambios en la entidad molecular durante las presentaciones regulatorias. Para permitir la coherencia en el uso de masas elementales, se incluye un diccionario de masas elementales con la herramienta de software como un archivo de texto de valores separados por comas (csv): Element_Mass.csv (Archivo de codificación suplementario 1). Del mismo modo, se incluye una lista curada de composiciones de glicanos que se ven típicamente en mAbs: Glycan.csv (Supplemental Coding File 2). Ambos archivos se guardan en la misma ubicación de carpeta que una aplicación ejecutable y pueden ser modificados por el usuario para usar una lista de masa elemental específica o una biblioteca de glicanos.
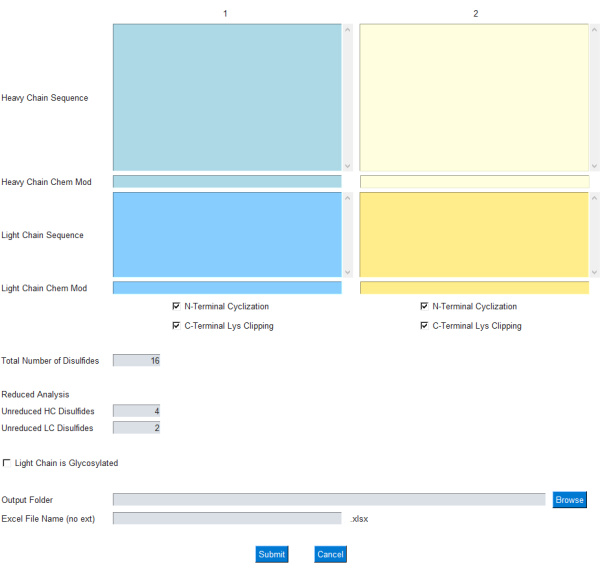
Figura 1: Interfaz gráfica de usuario para la aplicación mAbScale. El módulo GUI permite al usuario especificar la entrada para el cálculo de masa. El usuario introduce secuencias de aminoácidos de una sola letra para las cadenas de anticuerpos ligeros y pesados. Las modificaciones comunes para la ciclación del terminal N de cadena pesada y el recorte de lisina del terminal C se incluyen como casillas de verificación. Las fórmulas químicas/composiciones elementales se pueden sumar/restar a través del cuadro de texto respectivo de Chem Mod . Haga clic aquí para ver una versión más grande de esta figura.
