The photobleaching pre-treatment step can be added to a standard immunofluorescence protocol immediately prior to antigen retrieval and immunostaining (Figure 1A). Assembly of the photobleaching apparatus can also be performed using various, inexpensive, off-the-shelf components (Figure 1B). The emission spectrum of white phosphor LEDs covers a wide range of wavelengths which makes them suitable for broad-range photobleaching, agreeing with previous reports (Figure 1C)5,11. After 48 h photobleaching, the intensity of autofluorescent speckles that resemble lipofuscin as well as general background fluorescence was reduced considerably in both emission wavelengths in an unstained section of FTLD-T (Figure 1D). To demonstrated the efficacy of photobleaching, we stained for hyperphosphorylated tau to visualize pathological tau inclusions in a case of FTLD-T using two different secondary antibodies. The distinct shape of tau inclusions and the use of two chromophores for labeling also allows us to confidently distinguish autofluorescent features from the intended targets to validate the technique.
In the composite images at lower magnification, the morphology of the tau-positive inclusions, stained yellow from the combination of Alexa 488 and Texas Red channels, consists of ring-shaped collections of short cell processes that are referred to as 'astrocytic plaques' (Figure 2a-c). This morphology is characteristic of corticobasal degeneration (CBD)12,13, which agrees with the pathology report of this case. In the untreated sample, numerous structures are present in the composite image that did not resemble tau inclusions, and showed fluorescence primarily in the red channel, suggesting that it is autofluorescence rather than intended antibody staining. A noticeable level of background fluorescence is also visible in this channel throughout the field of view (Figure 2a). These autofluorescent features are removed and the overall image appears much cleaner when samples were treated with photobleaching (PB) and the chemical quencher (Figure 2b-c).
We then quantified the fluorescence differences in these samples by profiling a smaller region in each sample. A single astrocytic plaque is presented in each treatment condition for comparison (Figure 2d-r). In the untreated sample, background fluorescence is present in all three channels, but secondary antibody fluorescence for both Alexa 488 and Texas Red antibodies was relatively high (Fig 2h). However, the autofluorescent structures present in the untreated sample had fluorescence intensities in the Texas Red channel comparable to the Texas Red secondary antibody that stained for tau (Figure 2h). If the target protein did not have a distinct, predictable morphology such as tau in our case, the autofluorescence in the tissue would have rendered the image uninterpretable.
In contrast, when the photobleaching pre-treatment was applied prior to immunostaining, background fluorescence in the Alexa 488 and Texas Red channels was significantly reduced compared to the untreated sample, and the fluorescence of immunostained tau remained unaffected (Figure 2i-m). The commercial chemical quencher quenched the autofluorescence in the Alexa 488 channel just as effectively as photobleaching. It also suppressed DAPI background fluorescence, which was not affected by the 48 h photobleaching treatment (Figure 2m). However, the chemical quencher also reduced the intensity of the Texas Red secondary and DAPI signals, suggesting a certain degree of counterproductive quenching (Figure 2n-r).
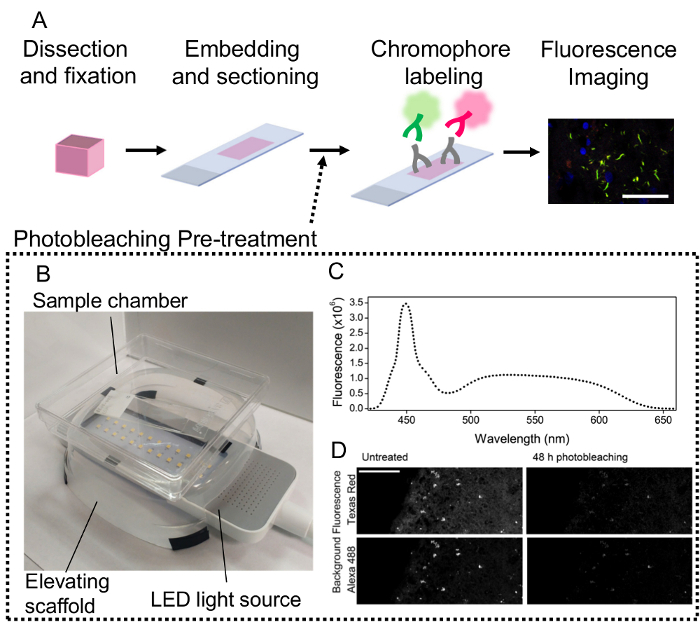
Figure 1: Application of photobleaching pre-treatment in a standard immunofluorescence workflow. A) Simplified schematic of the standard immunofluorescence protocol from tissue acquisition to imaging. Application of primary and secondary fluorescent antibodies is represented by cartoons. A representative microscope image is produced. Scale bar = 100 µm. B) A representative photobleaching apparatus constructed using off-the-shelf components (reflective lid is not shown). C) Emission spectrum of white LED array. A narrow emission peak at 450 nm and a broad peak at 550 nm are observed. D) Effect of photobleaching on endogenous background fluorescence of FTLD-T tissue at Alexa 488 (493-570 nm) and Texas Red (601-635 nm) emission wavelengths. Autofluorescent speckles resembling lipofuscin are visibly reduced after 48 h photobleaching. Scale bar = 100 µm. Please click here to view a larger version of this figure.
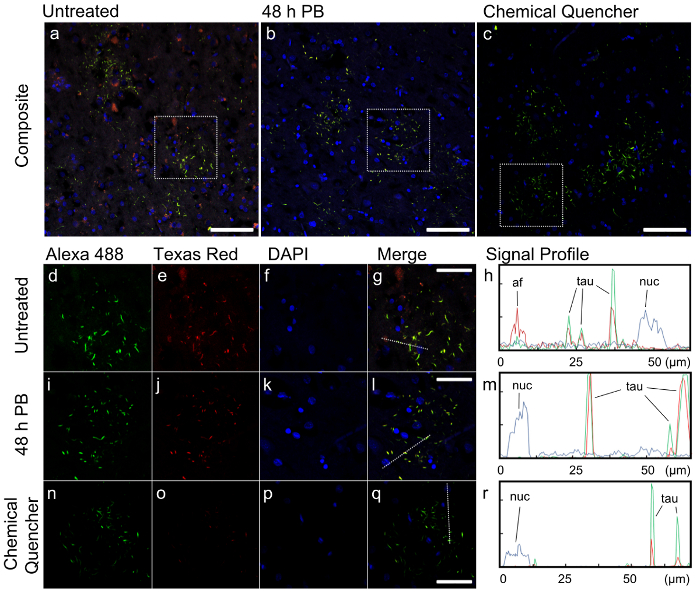
Figure 2:Effect of autofluorescence removal on image quality of a case of FTLD-T tissue with anti-phosphorylated tau immunostaining. a-c) Low magnification, composite immunofluorescence images of representative fields of view in untreated (a), photobleached for 48 h (b) and chemical quencher treated (c) samples. Colors represent fluorescence in the following channels via excitation by their respective light sources: Alexa 488 (green): λex = 488 nm (argon laser) λem = 493-570 nm; Texas Red (red): λex = 561 nm (DPSS 561 nm laser), λem = 601-635 nm; DAPI (blue): λex = 405 nm (Diode 405 laser), λem = 410-507 nm. Scale bar = 100 µm. d-r) Higher magnification images of the dotted regions in untreated (d-g), photobleached (i-l) and chemical quencher treated (n-q) samples, with separate fluorescence channels, merged image, and quantified fluorescence signal profiles. Dotted lines in the merged channels (g, l, q) represent the line on which signal profiles (h, m, r) were generated. Scale bar = 50 µm. Autofluorescent particles (af), immunolabeled tau fluorescence (tau) and nucleus signal (nuc) are indicated. Please click here to view a larger version of this figure.
